
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Учебное пособие: Общая и неорганическая химия
Учебное пособие: Общая и неорганическая химия
Квантово-механическая модель атома. Квантовые числа. Атомные орбитали. Порядок заполнения орбиталей электронамиТеория строения атома основана на законах, описывающих движение микрочастиц (электронов, атомов, молекул) и их систем (например, кристаллов). Массы и размеры микрочастиц чрезвычайно малы по сравнению с массами и размерами макроскопических тел. Поэтому свойства и закономерности движения отдельных микрочастиц отличаются от свойств и закономерностей движения макроскопических тел, изучаемых классической физикой. Движение и взаимодействие микрочастиц описывает квантовая механика, которая основывается на представлении о квантовании энергии, волновом характере движения микрочастиц и вероятностном (статистическом) методе описания микрообъектов.
Примерно в начале XX в. исследования явлений (фотоэффект, атомные спектры) привели к выводу, что энергия распространяется и передаётся, поглощается и испускается не непрерывно, а дискретно, отдельными порциями – квантами. Энергия системы микрочастиц также может принимать определённые значения, которые являются кратными частицами квантов.
Предположение
о квантовании
энергии впервые
было высказано
М. Планком в
1900 г. и было обосновано
Эйнштейном
в 1905 г.: энергия
кванта
![]() зависит от
частоты излучения
зависит от
частоты излучения
![]() :
:
![]() ,
,
где (1)
![]() – постоянная
Планка (
– постоянная
Планка (![]() )
)
Частота
колебаний
![]() и длина волны
и длина волны
![]() связаны соотношением:
связаны соотношением:
![]() ,
,
где
![]() – скорость
света.
– скорость
света.
Согласно
соотношению
(1), чем меньше
![]() ,
тем больше
энергия кванта
,
тем больше
энергия кванта
![]() и наоборот.
Таким образом,
ультрафиолетовые
и рентгеновские
лучи обладают
большей энергией,
чем скажем
радиоволны
и инфракрасные
лучи. Для описания
электромагнитного
излучения
привлекают
как волновые,
так и корпускулярные
представления:
с одной стороны
монохроматическое
излучение
распространяется
как волна и
характеризуется
длиной волны
и наоборот.
Таким образом,
ультрафиолетовые
и рентгеновские
лучи обладают
большей энергией,
чем скажем
радиоволны
и инфракрасные
лучи. Для описания
электромагнитного
излучения
привлекают
как волновые,
так и корпускулярные
представления:
с одной стороны
монохроматическое
излучение
распространяется
как волна и
характеризуется
длиной волны
![]() ,
с другой стороны
оно состоит
из микрочастиц
– фотонов,
переносящих
кванты энергии.
,
с другой стороны
оно состоит
из микрочастиц
– фотонов,
переносящих
кванты энергии.
Явление
дифракции
электромагнитного
излучения
доказывает
его волновую
природу. В то
же время электромагнитное
излучение
обладает энергией,
массой, производит
давление. Так,
вычислено, что
за 1 год масса
Солнца уменьшается
за счёт излучения
на
![]() .
.
В 1924 г. Луи де
Бройль предложил
распространить
корпускулярно-волновые
представления
на все микрочастицы,
т.е. движение
любой микрочастицы
рассматривать
как волновой
процесс. Математически
это выражается
соотношением
де Бройля, согласно
которому частице
массой
![]() ,
движущейся
со скоростью
,
движущейся
со скоростью
![]() ,
соответствует
волна длиной
,
соответствует
волна длиной
![]() :
:
![]() ,
(2)
,
(2)
![]() – импульс
частицы.
– импульс
частицы.
Гипотеза де Бройля была экспериментально подтверждена обнаружением дифракционного и интерферентного эффектов потока электронов.
Согласно
соотношению
(2) движению
электрона (![]() ,
,
![]() )
отвечает волна
длиной
)
отвечает волна
длиной
![]() ,
т.е. её длина
соизмерима
с размерами
атомов.
,
т.е. её длина
соизмерима
с размерами
атомов.
В 1925 г. Шрёдингер
предположил,
что состояние
движения электрона
в атоме должно
описываться
уравнением
стоячей электромагнитной
волны. Он получил
уравнение,
которое энергию
электрона
связывает с
пространством
Декартовых
координат и
так называемой
волновой функцией
![]() ,
которая соответствует
амплитуде 3-х
мерного волнового
процесса:
,
которая соответствует
амплитуде 3-х
мерного волнового
процесса:
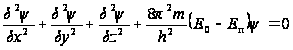 ,
где
,
где
![]() – полная
энергия электрона
– полная
энергия электрона
![]() – потенциальная
энергия электрона
– потенциальная
энергия электрона
![]() – вторая
частная производная
– вторая
частная производная
![]()
Уравнение
Шредингера
позволяет найти
волновую функцию
![]() как функцию
координат.
Физический
смысл волновой
функции в том,
что квадрат
её модуля определяет
вероятность
нахождения
электрона в
элементарном
объёме
как функцию
координат.
Физический
смысл волновой
функции в том,
что квадрат
её модуля определяет
вероятность
нахождения
электрона в
элементарном
объёме
![]() ,
т.е. характеризует
электронную
плотность.Т.
к. электрон
обладает свойствами
волны и частицы,
мы не можем
определить
его положение
в пространстве
в определённый
момент времени.
Электрон размазан,
т.е. делокализирован
в пространстве
атома. В этом
заключается
принцип Гейзенберга.
,
т.е. характеризует
электронную
плотность.Т.
к. электрон
обладает свойствами
волны и частицы,
мы не можем
определить
его положение
в пространстве
в определённый
момент времени.
Электрон размазан,
т.е. делокализирован
в пространстве
атома. В этом
заключается
принцип Гейзенберга.
Микрочастица, так же как и волна не имеет одновременно точных значений координат и импульса. Это проявляется в том, что чем точнее определяется координаты частицы, тем неопределеннее её импульс, и наоборот. Поэтому мы говорим о максимально вероятном нахождении электрона в данном месте в определённый момент времени. Та область пространства, где >90% находится электрон называется атомной орбиталью. Уравнение Шредингера имеет множество решений, но физически осмысленное решение только в определённых условиях.
Для описания
стоячей волны,
образованной
в атоме движущимся
электроном,
т.е. для нахождения
волновой функции
![]() необходимы
квантовые
числа.
необходимы
квантовые
числа.
В 3-х мерном пространстве 4-мя квантовыми числами описывается состояние электрона:
Главное
квантовое число
![]() характеризует
удалённость
электрона от
ядра и определяет
его энергию
(чем больше
характеризует
удалённость
электрона от
ядра и определяет
его энергию
(чем больше
![]() ,
тем больше
энергия электрона
и тем меньше
энергия связи
с ядром).
,
тем больше
энергия электрона
и тем меньше
энергия связи
с ядром).
![]() принимает
целочисленные
значения от
1 до Ґ.
принимает
целочисленные
значения от
1 до Ґ.
Состояние
электрона
характеризующееся
различными
значениями
главного квантового
числа
![]() ,
называется
электронным
слоем (электронной
оболочкой,
энергетическим
уровнем). Они
обозначаются
цифрами 1, 2, 3, 4, 5, …
или соответственно
буквами K,
L, M, N,
O ….
,
называется
электронным
слоем (электронной
оболочкой,
энергетическим
уровнем). Они
обозначаются
цифрами 1, 2, 3, 4, 5, …
или соответственно
буквами K,
L, M, N,
O ….
Квантовое
состояние атома
с наименьшей
энергией –
основное состояние,
а с более высокой
– возбуждённое
состояние.
Переход электрона
с одного уровня
на другой
сопровождается
либо поглощением,
либо выделением
энергии:
![]() .
.
Побочное
квантовое
(орбитальное,
азимутальное)
число
![]() (принимает все
целочисленные
значения от
0 до (n-1)).
(принимает все
целочисленные
значения от
0 до (n-1)).
|
|
|
Орбиталь |
| 1 | 0 | 1s |
| 2 | 0,1 | 2s,2p |
| 3 | 0,1,2 | 3s,3p,3d |
Состояние
электрона
характеризующееся
различными
значениями
побочного
квантового
числа
![]() называется
энергетическим
подуровнем.
В пределах
каждого уровня
с увеличением
называется
энергетическим
подуровнем.
В пределах
каждого уровня
с увеличением
![]() ,
растёт энергия
орбитали.
,
растёт энергия
орбитали.
Каждому
значению
![]() соответствует
определённая
форма орбитали
(например, при
соответствует
определённая
форма орбитали
(например, при
![]() – это сфера,
центр которой
совпадает с
ядром).
– это сфера,
центр которой
совпадает с
ядром).
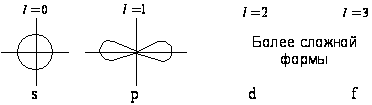
Магнитное
квантовое число
![]() характеризует
ориентацию
орбитали в
пространстве
(принимает все
целочисленные
значения от
-
характеризует
ориентацию
орбитали в
пространстве
(принимает все
целочисленные
значения от
-
![]() до +
до +![]() ).
).
Например,
для
![]()
![]() .
В пределах
каждого подуровня
орбиталь имеет
одинаковую
энергию.
.
В пределах
каждого подуровня
орбиталь имеет
одинаковую
энергию.
Спиновое
квантовое число
![]() характеризует
вращательный
момент, который
приобретает
электрон в
результате
собственного
вращения вокруг
своей оси (принимает
два значения:
характеризует
вращательный
момент, который
приобретает
электрон в
результате
собственного
вращения вокруг
своей оси (принимает
два значения:
![]() – вращение по
часовой стрелке,
– вращение по
часовой стрелке,
![]() – вращение
против часовой
стрелки).
– вращение
против часовой
стрелки).
Атомные орбитали заполняются электронами в соответствии с 3-мя принципами:
Принцип устойчивости (принцип min энергии): Каждая новая орбиталь заполняется только после того, как будут заполнены все предыдущие, т.е. более устойчивые (с min энергией) орбитали.
Энергия атомных орбиталей возрастает следующим образом:
![]()
Правило
Клечковского:
заполнение
электронами
атомных орбиталей
происходит
в соответствии
с увеличением
суммы главного
![]() и побочного
и побочного
![]() квантовых
чисел; если
квантовых
чисел; если
![]() одинакова, то
атомная орбиталь
заполняется
от больших
одинакова, то
атомная орбиталь
заполняется
от больших
![]() и меньших
и меньших
![]() к меньшим
к меньшим
![]() и большим
и большим
![]() .
.
|
|
|
|
Орбиталь |
| 1 | 0 | 1 | 1s |
| 2 | 0 | 2 | 2s |
| 1 | 3 | 2p | |
| 3 | 0 | 3 | 3s |
| 1 | 4 | 3p | |
| 2 | 5 | 3d | |
| 4 | 0 | 4 | 4s |
| 1 | 5 | 4p | |
| 2 | 6 | 4d | |
| 3 | 7 | 4f | |
| 5 | 0 | 5 | 5s |
| 1 | 6 | 5p | |
| 2 | 7 | 5d | |
| 3 | 8 | 5f | |
| 4 | 9 | 5g | |
| 6 | 0 | 6 | 6s |
Принцип Паули:
в атоме не может
быть 2 электрона,
у которых 4
одинаковых
квантовых
числа. Следовательно,
на 1-ой орбитали
могут находиться
не более 2-х
электронов,
отличающихся
друг от друга
значением
спинового
квантового
числа. Отсюда
следует, что
максимальное
количество
электронов
на энергетическом
уровне
![]() ,
на энергетическом
подуровне
,
на энергетическом
подуровне
![]() .
.
Пример:
![]()
![]()
![]()
Правило Хунда: электроны располагаются на орбиталях равной энергии таким образом, чтобы их суммарный спин был максимальный. Это означает, что первоначально электроны заполняют все свободные орбитали данного подуровня по 1-му, имея при этом параллельные спины, и только потом происходит заполнение этих орбиталей 2-ми электронами.
Пример:
![]()
![]() Px Py Pz
Px Py Pz
| ↑ | ↑ |
| ↑ | ↑ | ↑ |
| ↑↓ | ↑ | ↑ |
| K | L | M | ||||||||
|
|
1 | 2 | 3 | |||||||
|
|
0 | 0 | 1 | 0 | 1 | 2 | ||||
|
|
0 | 0 | -1 | 0 | +1 | 0 | -1 | 0 | +1 | |
|
|
↓↑ | ↓↑ | ↓↑ | ↓↑ | ↓↑ | ↓↑ | ↓↑ | ↓↑ | ↓↑ | |
|
|
|
|
|
|
|
|
|
|
||
Количество неспаренных электронов на внешнем уровне определяет валентность элемента, т.е. способность образовывать химические связи с другими атомами. В большинстве случаев, но не всегда.
![]() 5
5
![]()
![]() 4
4
![]()
4
![]() 3
3
![]()
3
![]() 2
2
![]()
2
![]() 1
1
![]()
1
![]()
Периодический закон (1869 г): свойства простых тел, а также свойства и формы соединений элементов находятся в периодической зависимости от величины атомных весов элементов.
До появления сведений о сложном строении атома основной характеристикой элемента служил атомный вес (относительная атомная масса). Развитие теории строения атома привело к установлению того факта, что главной характеристикой атома является положительный заряд ядра.
В современной формулировке периодический закон звучит: свойства химических элементов, а также формулы и свойства образуемых ими соединений находятся в периодической зависимости от величины заряда ядер их атомов.
Физической основой структуры периодической системы элементов служит определённая последовательность формирования электронных конфигураций атомов по мере роста порядкового номера элемента.
В зависимости от того, какой энергетический подуровень заполняется электронами последним, различают 4 типа элементов:
![]() – элементы
(последним
заполняется
– элементы
(последним
заполняется
![]() -подуровень
внешнего
энергетического
уровня)
-подуровень
внешнего
энергетического
уровня)
![]() – элементы
(последним
заполняется
– элементы
(последним
заполняется
![]() -подуровень
внешнего
энергетического
уровня)
-подуровень
внешнего
энергетического
уровня)
![]() – элементы
(последним
заполняется
– элементы
(последним
заполняется
![]() -подуровень
предпоследнего
энергетического
уровня)
-подуровень
предпоследнего
энергетического
уровня)
![]() – элементы
(последним
заполняется
– элементы
(последним
заполняется
![]() -подуровень
3-го снаружи
энергетического
уровня).
-подуровень
3-го снаружи
энергетического
уровня).
Горизонтально
располагаются
периоды –
последовательный
ряд элементов,
электронная
конфигурация
внешнего
энергетического
уровня которых
изменяется
от
![]() до
до
![]() .
Номер периода
совпадает со
значением
главного квантового
числа
.
Номер периода
совпадает со
значением
главного квантового
числа
![]() внешнего
энергетического
уровня.
внешнего
энергетического
уровня.
Вертикально
располагаются
группы – элементы
имеющие сходное
электронное
строение. У
элементов
главной подгруппы
последним
заполняется
![]() и
и
![]() подуровни
внешнего
энергетического
уровня, у элементов
побочной подгруппы
происходит
заполнение
внутренних
подуровни
внешнего
энергетического
уровня, у элементов
побочной подгруппы
происходит
заполнение
внутренних
![]() и
и
![]() подуровней.
Одинаковый
номер группы,
как правило,
определяет
число электронов,
которое может
участвовать
в образовании
химических
связей.
подуровней.
Одинаковый
номер группы,
как правило,
определяет
число электронов,
которое может
участвовать
в образовании
химических
связей.
Строение многоэлектронных атомов. Принцип наименьшей энергии, принцип Паули, правило Гунда, правило Клечковского. Электронные формулы
Число электронов, которые могут находиться на одном энергетическом уровне, определяется формулой 2n2, где n – номер уровня. Максимальное заполнение первых четырех энергетических уровней: для первого уровня – 2 электрона, для второго – 8, для третьего – 18, для четвертого – 32 электрона. Максимально возможное заполнение электронами более высоких энергетических уровней, в атомах известных элементов не достигнуто.
Квантово-механические расчеты показывают, что в многоэлектронных энергия электронов одного уровня неодинакова; электроны заполняют атомные орбитали разных видов и имеют разную энергию. Каждый энергетический уровень, кроме первого, расщепляется на такое число энергетических подуровней, сколько видов орбиталей включает этот уровень. Второй энергетический уровень расщепляется на два подуровня (2s – и 2p-подуровни), третий энергетический уровень – на три подуровня (3s-, 3p- и 3d-подуровни).
Каждый s-подуровень содержит одну s орбиталь, каждый р-подуровень – три р-орбитали, каждый d-подуровень семь f-орбиталей.
Закономерность заполнения электронных оболочек атомов определяется принципом запрета, установленным в 1925 г швейцарским физиком Паули (принцип Паули):
В атоме не могут одновременно находиться два электрона с одинаковым набором четырех квантовых квантовых чисел (заполнение электронами орбиталей происходит следующим образом: сначала на каждой орбитали располагается по одному электрону, затем, после заполнения всех орбиталей происходит распределение вторых электронов с противоположным спином).
Используя понятия квантовые числа можно сказать, что:
Каждый электрон в атоме однозначно характеризуется своим набором четырех квантовых чисел - главного n, орбитальногоl, магнитного ml, и спинового ms.
Заселение электронами энергетических уровней, подуровней и атомных орбиталей подчиняется следующему правилу:
В невозбужденном атоме все электроны обладают наименьшей энергией (принцип наименьшей энергии).
Это означает, что каждый из электронов, заполняющих оболочку атома, занимает такую орбиталь, чтобы атом в целом имел минимальную энергию. Последовательно квантовое возрастание энергии подуровней происходит в следующем порядке: 1s - 2s -2р - 3s – 3р - 4s –3d - 4р - 5s -….
Такой порядок увеличения энергии подуровней определяет расположение эле Ментов в Периодической системе.
Заполнение атомных орбиталей внутри одного энергетического подуровня происходит в соответствии с правилом, сформулированным немецким физиком Ф. Хундом (1927г) (правило Хунда):
При данном значении квантового числа l (т.е. в пределах одного подуровня) в основном состоянии электроны располагаются таким образом, что значение суммарного спина атома максимально. Это означает, что на подуровне должно быть максимально возможное число неспаренных электронов.
Порядок возрастания энергии атомной орбитали в сложных атомах описывается правилом Клечковского: энергия атомной орбитали возрастает в соответствии с увеличением n +l главного и орбитального квантовых чисел. При одинаковом значении суммы энергия меньше у атомной орбитали с меньшим значением главного квантового числа.
Распределение электронов по различным атомным орбиталям называют электронной конфигурацией атома. Электронная конфигурация с наименьшей энергией соответствует основному состоянию атома, остальные конфигурации относятся к возбужденным состояниям.
Электронную конфигурацию атома изображают двумя способами – в виде электронных формул и электронно-графических диаграмм. При написании электронных формул используют главное и орбитальное квантовые числа. Подуровень обозначают с помощью главного квантового числа (цифрой) и орбитального квантового числа (соответствующей буквой). Число электронов на подуровне характеризует верхний индекс. Например. Для основного состоянии атома водорода электронная формула: 1s1.
Более полно строение электронных подуровней можно описать с помощью электронографических диаграмм, где распределение электронов по подуровням представляют в виде квантовых ячеек. Орбиталь в этом случае принято условно изображать квадратом, около которого проставлено обозначение подуровня. Подуровни на каждом уровне должны быть немного смещены по высоте, так как их энергия несколько различается. Электроны обозначают стрелками или Ї в зависимости от знака спинового квантового числа.
С учетом структуры электронных конфигураций атомов все известные элементы в соответствии со значением орбитального квантового числа последнего заполняемого подуровня можно разбить на четыре группы: s –элементы, р-элементы, d-элементы, f-элементы.
Основные типы химической связи. Характеристики химической связи. Энергия связи. Длина связи
При образовании химической связи происходит перераспределение в пространстве электронных плотностей, первоначально принадлежавших разным атомам. Поскольку наименее прочно связаны с ядром электроны внешнего уровня, то этим электронам принадлежит главная роль в образовании химической связи. Количество химических связей, образованных данным атомом в соединении, называют валентностью. Электроны, принимающие участие в образовании химической связи, называются валентными: у s- и р элементов — это внешние электроны, у d- элементов — внешние (последние) s-электроны и предпоследние d-электроны. С энергетической точки зрения наиболее устойчивым является атом, на внешнем уровне которого содержится максимальное число электронов (2 и 8 электронов). Такой уровень называют завершенным. Завершенные уровни отличаются большой прочностью и характерны для атомов благородных газов, поэтому при обычных условиях они находятся в состоянии химически инертного одноатомного газа.
У атомов других элементов внешние энергетические уровни незавершенные. В процессе хим реакции осуществляется завершение внешних уровней, что достигается либо присоединением, либо отдачей электронов, а также образованием общих электронных пар. Эти способы приводят к образованию двух основных типов связи: ковалентной и ионной. Таким образом, при образовании молекулы каждый атом стремится приобрести устойчивую внешнюю электронную оболочку: либо двухэлектронную (дублет), либо восьми-злектромную (октет). Эта закономерность положена в основу теории образования химической связи. Образование химической связи за счет завершения внешних уровней в образующих связь атомах сопровождается выделением большого количества энергии, то есть возникновение химической связи всегда протекает экзотермически, поскольку оно приводит к появлению новых частиц (молекул), обладающих при обычных условиях большей устойчивостью, а следовательно, они меньшей энергией, чем у исходных. Одним из существенных показателей, определяющих какая связь образуется между атомами, является электроотрицательность, то есть способность атомом притягивать к себе электроны от других атомов. Электроотрицательность атомов элементов изменяется постепенно: в периодах периодической системы слева направо ее значение возрастает а в группах сверху вниз — уменьшается.
Химическая связь, осуществляемая за счет образования общих (связывающих) электронных пар, называется ковалентной.1) Разберем пример образования химической связи между атомами с одинаковой электроотрицательностью, например, молекулы водорода Н2 Образование химической связи в молекуле водорода можно представить в виде двух точек: Н- + -Н -> Н : Н или черточкой, которая символизирует пару электронов: H-H Ковалентная связь, образованная атомами с одинаковой электроотрицательностью называется неполярной. Такую связь образуют двухатомные молекулы, состоящие из атомов одного химического элемента: H 2, Cl 2 и др.2) Образование ковалентной связи между атомами, электроотрицательность которых различается незначительно. Ковалентная связь, образованная атомами с различной электроотрицательностью, называется полярной. При ковалентной полярной связи электронная плотность от общей пары электронов смещена к атому с большей электроотрицательностью. Примерами могут служить молекулы Н2О, NH3, H2S, CH3Cl. Ковалентная (полярная и неполярная) связь в наших примерах образовалась за счет неспаренных электронов связывающихся атомов. Такой механизм образования ковалентной связи называется обменным. Другой механизм образования ковалентной связи — донорно-акцепторный. В этом случае связь возникает за счет двух спаренных электронов одного атома (донора) и свободной орбитали другого атома (акцептор). Хорошо известный пример — образование иона аммония: Н++:NH 3 -> [ Н : NH3 | + <=====> NH4+ акцептор донор ион аммония электронов. При образовании иона аммония электронная пара азота становится общей для атомов N и Н, то есть возникает четвертая связь, которая не отличается от остальных трех. Их изображают одинаково:
Н+
H-N-H
Н
Ионная связь возникает между атомами, электроотрицательность которых резко различается Рассмотрим способ образования на примере хлорида натрия NaCl. Электронную конфигурацию атомов натрия и хлора можно представить: 11 Na ls2 2s2 2p 6 3s1; 17 Cl ls2 2p 6 Зs2 3р5 Как это атомы с незавершенными энергетическими уровнями. Очевидно, для их завершения атому натрия легче отдать один электрон, чем присоединить семь, а атому хлора легче присоединить один электрон, чем отдать семь. При химическом взаимодействии атом натрия полностью отдает один электрон, а атом хлора принимает его. Схематично это можно записать так: Na. — l е —> Na+ ион натрия, устойчивая восьмиэлектронная 1s2 2s2 2p6 оболочка за счет второго энергетического уровня. :Cl + 1е -->.Cl - ион хлора, устойчивая восьмиэлектронная оболочка. Между ионами Na+ и Cl- возникают силы электростатического притяжения, в результате чего образуется соединение.
Химическая связь, осуществляемая за счет электростатического притяжения между ионами, называется ионной связью. Соединения, образованные путем притяжения ионов называются ионными. Ионные соединения состоят из отдельных молекул только в парообразном состоянии. В твердом (кристаллическом) состоянии ионные соединения состоят из закономерно расположенных положительных и отрицательных ионов. Молекулы в этом случае отсутствуют. Ионные соединения образуют резко различные по величине электроотрицательности элементы главных подгрупп I и II групп и главных подгрупп VI и VII групп. Ионных соединений сравнительно немного. Например неорганические соли: NH4Cl (ион аммония NH4 + и ион хлора Cl-), а также солеобразные органические соединения: алкоголяты соли карбоновых кислот, соли аминов Неполярная ковалентная связь и ионная связь — два предельных случая распределения электронной плотности. Неполярной связи отвечает равномерное распределение связующего двух электронного облака между одинаковыми атомами. Наоборот, при ионной связи связующие электронное облако практически полностью принадлежит одному из атомов. В большинстве же соединений химические связи оказывают промежуточными между этими видами связи, то есть в них осуществляется полярная ковалентная связь.
Металлическая связь существует в металлах в твердом в жидком состоянии. В соответствии с положением в периодической системе атомы металлов имеют небольшое число валентных электронов (1-3 электрона) и низкую энергию ионизации (отрыва электрона). Поэтому валентные электроны слабо удерживаются в атоме, легко отрываются и имеют возможность перемещаться по всему кристаллу. В узлах кристаллической решетки металлов находятся свободные атомы, положительно заряженные коны, а часть валентных электронов, свободно перемещаясь в объеме кристаллической решетки, образует «электронный газ», обеспечивающий связь между атомами металла. Связь, которую осуществляют относительно свободные электроны между ионами металлов в кристаллической решетке, называется металлической связью. Металлическая связь возникает за счет обобществления атомами валентных электронов. Однако между этими видами связи есть существенное различие. Электроны, осуществляющие ковалентную связь, в основном пребывают в непосредственной близости от двух соединенных атомов. В случае металлической связи электроны, осуществляющие связь, перемещаются по всему куску металла. Этим определяются общие признаки металлов: металлический блеск, хорошая проводимость теплоты и электричества, ковкость, пластичность и т. д. Общим химическим свойством металлов является их относительно высокая восстановительная способность.
Водородные связи могут образовываться между атомом водорода, связанным с атомом электроотрицательного элемента, и электроотрицательным элементом, имеющим свободную пару электронов(О,F,N). Водородная связь обусловлена электростатическим притяжением, которому способствуют малые размеры атома водорода, и отчасти, донорно-акцепторным взаимодействием. Водородная связь может быть межмолекулярной и внутримолекулярной. Связи 0-Н имеют выраженный полярный характер: Водородная связь гораздо более слабая, чем ионная или ковалентная, но более сильная, чем межмолекулярное взаимодействие. Водородные связи обуславливают некоторые физические свойства веществ (например, высокие температуры кипения). Особенно распространены водородные связи в молекулах белков, нуклеиновых кислот и других биологически важных соединений, обеспечивая им определенную пространственную структуру (организацию).
Энергия связи(Eсв). Кол-во энергии, выделяющейся при образовании химической связи, называется энергией химической связи[кДж/моль]. Для многоатомных соединений принимают среднее её значение. Чем больше Eсв тем устойчивее молекула.
Длина связи(lсв). Расстояние между ядрами в соединении. Чем больше длина связи — тем меньше энергия связи.
Метод валентных связей.
А) химическая связь между двумя атомами возникает как результат перекрытия АО с образованием электронных пар.
Б) атомы вступающие в химическую связь, обмениваются между собой электронами, которые образуют связывающие пары. Энергия обмена электронами между атомами(энергия притяжения атомов) вносит основной вклад в энергию химической связи. Дополнительный вклад в энергию связи дают кулоновские силы взаимодействия частиц.
В) в соответствии с принципом Паули химическая связь образуется лишь при взаимодействии электронов с разными спинами.
Г)характеристики химической связи(энергия, длина, полярность) определяются типом перекрывающихся АО.
Метод валентных связей. Ковалентная связь направлена в сторону максимального перекрывания АО реагирующих атомов.
Валентность. Способность атома присоединять или замещать определённое число других атомов с образованием химических связей.
При переходе в возбуждённое состояние, один из спаренных электронов переходит в свободную орбиталь той же оболочки.
Донорно-акцепторный механизм: образуется общая электронная пара за счёт неподелённой пары электронов одного атома и вакантной орбитали другого атома.
Метод молекулярных орбиталей. Электроны в молекуле распределены по МО, которые подобно АО характеризуются определённой энергией и формой. МО охватывают всю молекулу. Молекула рассматривается как единая система.
1. Число МО равно общему числу АО, из которых комбинируется МО.
2. Энергия одних МО оказывается выше, других — ниже энергии исходных АО. Средняя энергия МО, полученная из набора АО, приблизительно совпадает с средней энергией этих АО.
3. Электроны заполняют МО, как и АО, в порядке возрастания энергии, при это соблюдается принцип запрета Паули и правило Гунда.
4. Наиболее эффективно комбинируются АО с теми АО которые характеризуются сопоставимыми энергиями и соответствующей симметрией.
5. Как и в методе ВС, прочность связи в методе МО пропорциональна степени перекрывания атомных орбиталей.
Порядок и энергия связи. Порядок связи n=(Nсв-Nр)/2. Nсв — число e на связывающих молекулярных орбиталях, Nр — число e на разрыхляющих молекулярных орбиталях.
Если Nсв = Nр, то n=0 и молекула не образуется. С увеличением n в однотипных молекулах растёт энергия связи. В отличии от метода АО, в методе МО допускается, что связь может быть образована одним электроном.
Комплексные соеденения. Сложные соединения у которых имеются ковалентные связи, образованные по донорно акцепторному механизму
4. Ковалентная (неполярная, полярная) связь. Механизмы образования ковалентной связи
При помощи химической связи атомы элементов в составе веществ удерживаются друг возле друга. Тип химической связи зависит от распределения в молекуле электронной плотности.
Химическая связь – взаимное сцепление атомов в молекуле и кристаллической решетке под воздействием электрических сил притяжения между атомами. Атом на внешнем энергетическом уровне способен содержать от одного до восьми электронов. Валентные электроны – электроны предвнешнего, внешнего электронных слоев, участвующие в химической связи. Валентность – свойство атомов элемента образовывать химическую связь.
Ковалентная связь образуется за счет общих электронных пар, возникающих на внешних и предвнешних подуровнях связываемых атомов.
Общая электронная пара осуществляется через обменный или донорно-акцепторный механизм. Обменный механизм образования ковалентной связи – спаривание двух неспа-ренных электронов, принадлежащих различным атомам. Донорно-акцепторный механизм образования ковалетной связи – образование связи за счет пары электронов одного атома (донора) и вакантной орбитали другого атома (акцептора).
Есть две основные разновидности ковалентной связи: неполярная и полярная.
Ковалентная неполярная связь возникает между атомами неметалла одного химического элемента (O2, N2, Cl2) – электронное облако связи, образованное общей парой электронов, распределяется в пространстве симметрично по отношению к ядрам обоих атомов.
Ковалентная полярная связь возникает между атомами различных неметаллов (HCl, CO2, N2O) – электронное облако связи смещается к атому с большей электроотрицательностью.
Чем сильнее перекрываются электронные облака, тем прочнее ковалентная связь.
Электроотрицательность – способность атомов химического элемента оттягивать к себе общие электронные пары, участвующие в образовании химической связи.
Свойства ковалентной связи: 1) энергия; 2) длина; 3) насыщаемость; 4) направленность.
Длина связи – расстояние между ядрами атомов, образующих связь.
Энергия связи – количество энергии, необходимое для разрыва связи.
Насыщаемость – способность атомов образовывать определенное число ковалентных связей.
Направленность ковалентной связи – параметр, определяющий пространственную структуру молекул, их геометрию, форму.
Гибридизация – выравнивание орбиталей по форме и энергии. Существует несколько форм перекрывания электронных облаков с образованием ?-связей и ?-связей (?-связь намного прочнее ?-связи, ?-связь может быть только с ?-связью). Ковалентная связь - это связь, возникающая между атомами за счет образования общих электронных пар. В основе ее также лежит представление о приобретении атомами энергетически выгодной и устойчивой электронной конфигурации из 8 электронов (для атома водорода из 2). Такую конфигурацию атомы получают не путем отдачи или присоединения электронов как в ионной связи, а посредством образования общих электронных пар. Механизм образования такой связи может быть обменный или донорно-акцепторный.
К обменному механизму относят случаи, когда в образовании электронной пары от каждого атома участвует по одному электрону. Например водород: Н2 Н. +Н. →Н:Н или Н-Н. Связь возникает благодаря образованию общей электронной пары за счет объединения неспаренных электронов. У каждого атома есть по одному s –электрону. Атомы Н равноценны и пары одинаково принадлежат обоим атомам. По этому же принципу происходит образование общих электронных пар (перекрывание р-электронных облаков) при образовании молекулы Сl2. При образовании молекулы N2 Образуются 3 общие электронные пары. Перекрываются р-орбитали. Связь называется неполярная.
При образовании молекулы хлороводорода перекрывается орбиталь s-электрона водорода и орбиталь р-электрона хлора Н-Сl. Связывающая электронная пара смещена к атому хлора, в результате чего образуется диполь, который измеряется дипольным моментом. Связь называется полярная.
По донорно-акцепторному механизму происходит образование иона аммония. Донор (азот) имеет электронную пару, акцептор – (Н+) свободную орбиталь, которую пара электронная азота может занять. В ионе аммония три связи азота с водородом образованы по обменному механизму, а одна по донорно-акцепторному. Все 4 связи равноценны.
Ковалентные связи классифицируют не только по механизму образования общих электронных пар, соединяющих атомы, но и по способу перекрывания электронных орбиталей, по числу общих пар, а также по смещению их. По способу перекрывания – у (сигма s- s, s-р, р-р) р (р-р гантели перекрываются двумя местами). В молекуле азота между атомами существуют одна у-связь и две р-связи, которые находятся в двух взаимно перпендикулярных плоскостях.
По числу общих электронных пар различают: одинарные Н2, НСl; двойные С2Н4, СО2; тройные N2.
По степени смещенности: полярные и неполярные. Связь между атомами с одинаковой электроотрицательностью – неполярная, с разной – полярная.
Исследования ученых позволили сделать вывод, что химическая связь в молекуле водорода осуществляется путем образования пары электронов с противоположно направленными спинами. Каждый электрон занимает место в квантовых ячейках обоих атомов, т.е. движется в силовом поле, образованном двумя силовыми центрами – ядрами атомов водорода. Это представление о механизме образования химической связи было развито учеными Гейтлером и Лондоном на примере водорода.это было распространено и на более сложные молекулы. Разработанная на этой основе теория образования химической связи получила название метода валентных связей. Метод ВС дал теоретическое объяснение важнейших свойств ковалентной связи, позволил понять строение большого числа молекул. Хотя этот метод не оказался универсальным и в ряде случаев не в состоянии правильно описать структуру и свойства молекул – все же он сыграл большую роль в разработке квантово-механической теории химической связи и не потерял своего значение до настоящего времени. В основе метода ВС лежат следующие положения:
- ковалентная связь образуется двумя электронами с противоположно направленными спинами, причем эта электронная пара принадлежит двум атомам.
-ковалентная связь тем прочнее, чем в большей степени перекрываются взаимодействующие электронные облака.
Геометрическая форма s –орбитали сферическая, от центра к краям размазанная (более плотная у ядра, и менее- на краях). Орбитали р-электронов представляют собой гантели, направленные вдоль осей координат. Облака d –электронов имеют более сложную форму. Метод гибридизации орбиталей исходит из предположения, что при образовании молекул вместо исходных s-, р-, d-,f- орбиталей (облаков) образуются такие равноценные «смешанные» или гибридные электронные облака, которые вытянуты по направлению к соседним атомам, благодаря чему достигается более полное их перекрывание с электронными облаками других атомов. На гибридизацию затрачивается энергия, за то она окупается более полным перекрыванием. Получается более прочная молекула. Затраченная на гибридизацию энергия окупается энергией, выделяющейся при образовании связи. Пример –молекула метана.В результате перекрывания четырех гибридных sр3 орбиталей атома углерода и 4 s орбиталей 4-х атомов водорода, образуется тетраэдрическая модель молекулы метана с четырьмя у связями, под углом 1090. Если в молекуле гибридизуется 3-р орбитали, то sр2 гибридизация – молекула этилена, если 2 орбитали sр – гибридизция (ацетилен). У элементов 3 и последующих периодов в образовании гибридных облаков участвуют и d-электроны. В этом случае образуются 6 равноценных гибридных облака, вытянутых к вершинам октаэдра sр3 d2-гибридизация. Такую гибридизацию имеет центральный атом комплексного иона. Этим объясняется их октаэдрическая структура.
Ковалентная связь обладает направленностью. Область перекрывания располагается в определенном направлении по отношению к взаимодействующим атомам.
Характер распределения электронов по молекулярным орбиталям позволяет объяснить магнитные свойства частиц. Молекулы, суммарный спин которых равен нулю, проявляют диамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются против направления поля. Молекулы, суммарный спин которых отличен от нуля, проявляют парамагнитные свойства, т.е. во внешнем магнитном поле их собственные магнитные моменты ориентируются в направлении поля. Таким образом молекула Н2 диамагнитна.
Геометрическая форма молекул зависит от направленности химической связи. Ядра атомов молекул имеющих sр-гибридизацию атомных орбиталей расположены в одной плоскости, sр2 –направлены к вершинам треугольника, sр3 – к верщинам тетраэдра
Понятия о методах валентных связей и молекулярных орбиталей образования химических соединений
Химическая связь, осуществляемая за счет образования общих (связывающих) электронных пар, называется ковалентной.Для объяснения ковалентной связи используют 2 метода квантово-механического расчета: метод валентных связей (МВС) метод молекулярных орбиталей (ММО)
1916 году американский ученый Льюис высказал предположение о том, что химическая связь образуется за счет обобществления двух электронов.
При этом электронная оболочка атома стремится по строению к электронной оболочке благородного газа. В дальнейшем эти предположения послужили основой для развития метода валентных связей. В 1927году Гайтлером и Лондоном был выполнен теоретический расчет энергии двух атомов водорода в зависимости от расстояния между ними. Оказалось, что результаты расчета зависят от того, одинаковы или противоположны по знаку спины взаимодействующих электронов. При совпадающем направлении спинов сближение атомов приводит к непрерывному возрастанию энергии системы. При противоположно направленных спинах на энергетической кривой имеется минимум, т.е. образуется устойчивая система – молекула водорода Н2.
Образование химической связи между атомами водорода является результатом взаимопроникновения (перекрывания) электронных облаков. Вследствие этого перекрывания плотность отрицательного заряда в межъядерном пространстве возрастает, и положительно заряженные ядра притягиваются к этой области.
Представления о механизме образования молекулы водорода были распространены на более сложные молекулы. Разработанная на этой основе теория химической связи получила название метод валентных связей (метод ВС).
1) Ковалентная связь образуется двумя электронами с противоположно направленными спинами, причем эта электронная пара принадлежит двум атомам.
2) Ковалентная связь тем прочнее, чем в большей степени перекрываются электронные облака.
Комбинации двухэлектронных двухцентровых связей, отражающие электронную структуру молекулы, получили название валентных схем. Примеры построения валентных схем:
![]()
![]()
Ковалентная связь характеризуется длиной, энергией, полярностью, поляризуемостью и имеет определённую направленность в пространстве.
С увеличением
кратности
связи, длина
связи уменьшается:
![]() −
−![]() 0,154 нм
0,154 нм
![]() =
=![]() 0,134 нм
0,134 нм![]() ≡
≡![]() 0,12 нм
0,12 нм
Энергия связи – энергия, которую надо затратить, чтобы разорвать химическую связь. Тоже количество энергии выделяется при образовании химической связи. С увеличением кратности связи, энергия увеличивается. Энергия p-связи меньше энергии s-связи.
Свойства ковалентной связи: прочность, полярность, насыщаемость, направленность, гибридизация, кратность
1. Прочность ковалентной связи — это свойства характер длинной связи (межъядерное пространство) и энергии энергией связи.
2. Полярность ковалентной связи. В молекулах, содержащих ядра атомов одного и того же элемента, одна или несколько пар электронов в равной мере принадлежат обоим атомам, каждое ядро атома с одинаковой силой притягивает пару связывающих электронов. Такая связь называется неполярной ковалентной связью. Если пара электронов, образующих химическую связь, смещена к одному из ядер атомов, то связь называют полярной ковалентной связью.
3. Насыщаемость ковалентной связи — это способность атома участвовать только в определенном числе ковалентной связи, насыщаемость характеризует валентностью атома. Количественные меры валентности являются число не спаренных электронов у атома в основном и в возбужденном состоянии.
4. Направленность ковалентной связи. Наиболее прочные ковалентной связи образуются в направлении максимального перекрывания атомных орбиталей, то есть мерой направленности служит валентный угол.
5. Гибридизация ковалентной связи — при гибридизации происходит смещение атомных орбиталей, т.е. происходит выравнивание по энергии и по форме. Существует sp, sp2, sp3 —гибридизация. sp — форма молекулы линейная (угол 1800), sp2 — форма молекулы плоская треугольная (угол 1200), sp3 - форма тетраэдрическая (угол 109028).
6. Кратность ковалентной связи или делоколизация связи — Число связей, образующихся между атомами, называется кратностью (порядком) связи. С увеличением кратности (порядка) связи изменяется длина связи и ее энергия.
Типы химической связи. Ионная, металлическая связи
Металлическая связь При обычных условиях металлы, за исключением ртути Hg, существуют в виде кристаллов. Взаимодействие, удерживающее атомы металлов в едином кристалле, называется металлической связью.
Природа металлической связи подобна ковалентной связи: оба типа связи основаны на обобществлении валентных электронов. Однако в атомах металлов количество таких электронов меньше количества вакантных орбиталей. Электроны слабо удерживаются ядром. Поэтому они могут переходить из одной орбитали в другую. Стремясь принять более устойчивое состояние, а это структура инертного газа, атомы металлов довольно легко отдают валентные электронные электроны, превращаясь в положительно заряжённые ионы. Внутри этой решётки находятся валентные электроны, которые не принадлежат конкретно какому-то атому. Благодаря малым размерам электроны более или менее свободно перемещаются по всему объёму кристаллической решётки, поэтому возникает большое число многоцентрированных орбиталей. Электроны на этих орбиталях обобщены сразу несколькими атомами.
Благодаря свободному перемещению электронов металлы обладают высокой электрической проводимостью и теплопроводностью.
По прочности
металлическая
связь меньше
ковалентной
связи в 3-4 раза.
Металлическая
связь не имеет
определённой
направленности
в пространстве.
Электроны
сталкиваясь
с ионами образуют
нейтральные
частицы, которые
сразу теряют
электроны:
![]() .
Электронные
газы отражают
световые лучи.
.
Электронные
газы отражают
световые лучи.
В результате движения внутри решётки электроны способны переносить тепловую энергию от нагретых участков к ненагретым, этим объясняется теплопроводность.
Если приложить нагрузку к металлу, происходит деформация без разрушения решётки, металлам характерна ковкость, пластичность.
Ионная Химическая связь, осуществляемая за счет электростатического притяжения между ионами, называется ионной связью. Соединения, образованные путем притяжения ионов называются ионными. Ионные соединения состоят из отдельных молекул только в парообразном состоянии. В твердом (кристаллическом) состоянии ионные соединения состоят из закономерно расположенных положительных и отрицательных ионов. Молекулы в этом случае отсутствуют. Ионные соединения образуют резко различные по величине электроотрицательности элементы главных подгрупп I и II групп и главных подгрупп VI и VII групп. В зависимости от величины электроотрицательности все элементы делятся на:
электроположительные (элементы 1-3 группы)
электротрицательные (все остальные элементы)
Ионная связь образуется между элементами сильно отличающимися по электроотрицательности.
Ионных соединений сравнительно немного. Например неорганические соли: NH4Cl (ион аммония NH4 + и ион хлора Cl-), а также солеобразные органические соединения: алкоголяты соли карбоновых кислот, соли аминов Неполярная ковалентная связь и ионная связь — два предельных случая распределения электронной плотности.
Неполярной связи отвечает равномерное распределение связующего двух электронного облака между одинаковыми атомами. Наоборот, при ионной связи связующие электронное облако практически полностью принадлежит одному из атомов.
В большинстве же соединений химические связи оказывают промежуточными между этими видами связи, то есть в них осуществляется полярная ковалентная связь.
Потенциал ионизации – энергия, которую необходимо затратить для удаления 1-го электрона с внешней орбитали, при этом атом переходит из нейтрального в положительно заряженный ион (катион).
Чем меньше потенциал ионизации, тем легче атом теряет электроны, тем сильнее выражены у электрона металлические свойства. Потенциал ионизации растет в пределах периода слева направо, уменьшается сверху вниз.
Ионная связь образуется за счет перехода одного или нескольких электронов от одного атома на внешнюю оболочку другого атома.
Атом, отдавший электрон становится положительно заряженным, а получивший – отрицательно заряженный Связь между разноименными ионами осуществляется за счет сил электростатического притяжения.
Образование ионной связи происходит по октаэдрическому правилу. Согласно этому правилу атом принимает, теряет или разделяет электроны таким образом, чтобы электронное облако для него соответствовало ближайшему инертному газу.
Основные виды взаимодействия молекул. Силы межмолекулярного взаимодействия. Водородная связь
Межмолекулярные взаимодействия, взаимодействия молекул между собой, не приводящее к разрыву или образованию новых химических связей. Межмолекулярные взаимодействия определяют отличие реальных газов от идеальных, существование жидкостей и молекулярных кристаллов. От межмолекулярных взаимодействий зависят многие структурные, спектральные, термодинамические, теплофизические и другие свойства веществ. Появление понятия межмолекулярные взаимодействия связано с именем Й. Д. Ван-дер-Ваальса, который для объяснения свойств реальных газов и жидкостей предложил в 1873 уравнение состояния, учитывающее межмолекулярные взаимодействия. Поэтому силы межмолекулярного взаимодействия часто называют ван-дер-ваальсовыми.
Виды межмолекулярных взаимодействийОснову межмолекулярных взаимодействий составляют кулоновские силы взаимодействия между электронами и ядрами одной молекулы и ядрами и электронами другой. В экспериментально определяемых свойствах вещества проявляется усредненное взаимодействие, которое зависит от расстояния R между молекулами, их взаимной ориентации, строения и физических характеристик (дипольного момента, поляризуемости и др.). При больших R, значительно превосходящих линейные размеры l самих молекул, вследствие чего электронные оболочки молекул не перекрываются, силы межмолекулярного взаимодействия можно достаточно обоснованно подразделить на три вида - электростатические, поляризационные (индукционные) и дисперсионные. Электростатические силы иногда называют ориентационными, однако это неточно, поскольку взаимная ориентация молекул может обусловливаться также и поляризационными силами, если молекулы анизотропны.
При малых расстояниях между молекулами (R ~ l) различать отдельные виды межмолекулярных взаимодействий можно лишь приближенно, при этом, помимо названных трех видов, выделяют еще два, связанные с перекрыванием электронных оболочек, - обменное взаимодействие и взаимодействия, обязанные переносу электронного заряда. Несмотря на некоторую условность, такое деление в каждом конкретном случае позволяет объяснять природу межмолекулярного взаимодействия и рассчитать его энергию.
Водородная связь Водородные связи могут образовываться между атомом водорода, связанным с атомом электроотрицательного элемента, и электроотрицательным элементом, имеющим свободную пару электронов(О,F,N). Водородная связь обусловлена электростатическим притяжением, которому способствуют малые размеры атома водорода, и отчасти, донорно-акцепторным взаимодействием. Водородная связь может быть межмолекулярной и внутримолекулярной. Связи 0-Н имеют выраженный полярный характер: Водородная связь гораздо более слабая, чем ионная или ковалентная, но более сильная, чем межмолекулярное взаимодействие. Водородные связи обуславливают некоторые физические свойства веществ (например, высокие температуры кипения). Особенно распространены водородные связи в молекулах белков, нуклеиновых кислот и других биологически важных соединений, обеспечивая им определенную пространственную структуру (организацию).
В 80-х годах XIX в. М.А. Ильинский и Н.Н. Бекетов установили, что атом водорода, соединенный с атомом фтора, кислорода или азота, способен образовывать еще одну дополнительную связь – то есть некоторые водородосодер-жащие группы атомов образуют химическую связь, электроотрицательные атомы которой входят в состав молекулы. Этот вид связи получил название водородная связь.
Водородная связь – взаимодействие между двумя электроотрицательными атомами одной или нескольких разных молекул при помощи атома водорода: А—Н...В (чертой обозначена ковалентная связь, тремя точками – водородная связь).Для водородной связи характерно электростатическое притяжение водорода (несущего положительный заряд ?+) к атому электроотрицательного элемента, имеющего отрицательный заряд ?-. Чаще всего она слабее ковалентной, но сильнее обычного притяжения молекул друг к другу в твердых и жидких веществах.Водородная связь отличается от межмолекулярных взаимодействий тем, что обладает свойствами направленности и насыщаемости.Водородная связь считается разновидностью ковалентной химической связи. Описывается при помощи метода молекулярных орбита-лей в виде трехцентровой двухэлектронной связи.Признак наличия водородной связи – расстояние между атомом водорода и другим атомом, ее образующим, меньше, чем общая сумма радиусов этих атомов.
Чаще встречаются несимметричные водородные связи (расстояние Н...В>А—В ), редко – симметричные (HF ).Угол между атомами А—Н...В ~180o.Водородная связь присутствует во многих химических соединениях. Образуется между наиболее электроотрицательными элементами (фтор, азот, кислород), реже – в некоторых других (хлор, сера).Наиболее прочные водородные связи имеются в воде, фтороводороде, кислородсодержащих неорганических кислотах, карбоновых кислотах, фенолах, спиртах, аммиаке, аминах.
При кристаллизации водородные связи сохраняются. Кристаллические решетки водородных связей:
1) цепи (метанол);2) плоские двухмерные слои (борная кислота);3) пространственные трехмерные сетки (лед).Внутримолекулярная водородная связь – водородная связь, объединяющая части одной молекулы.
Межмолекулярная водородная связь – водородная связь, образующаяся между атомом водорода одной молекулы и атомом неметалла другой молекулы.
Цепные реакции. Механизм протекания цепных реакций
Существуют химические реакции, в которых взаимодействие между компонентами происходит довольно просто. Существует весьма обширная группа реакций, протекающих сложно. В этих реакциях каждый элементарный этап связан с предыдущим, без выполнения которого дальнейшая реакция невозможна. В таких реакциях образование продукта реакции являет собой результат цепи элементарных этапов реакции, что называется цепными реакциями, которые проходят при участии активных центров – атомов, ионов или радикалов (осколков молекул). Радикал – осколок молекулы, имеющий неспаренные электроны и проявляющий высокую реакционную активность (H, Cl, O, OH, CH3).
При взаимодействии активных центров с молекулами исходных компонентов происходит образование продуктов реакции и новых активных частиц, способствующих новому этапу взаимодействия. Активные центры способствуют и создают цепи последовательных превращений веществ.
В качестве примера цепной реакции можно привести реакцию синтеза хлористого водорода:
Эту реакцию провоцирует свет. Молекула хлора поглощает квант лучистой энергии hv и приходит в возбуждение, то есть атом в ней начинает энергично колебаться. Когда энергия колебаний превышает энергию связи, то происходит распад молекулы (фотохимическая диссоциация ):
Обрыв цепи – окончание цепи, характеризующееся соударением двух активных частиц и одной неактивной, результатом которой является образование молекулы и унос выделившейся энергии неактивной частицей.Цепные реакции делятся на: 1) неразветвленные цепные реакции;2) разветвленные цепные реакции.Неразветвленная цепная реакция характеризуется тем, что при каждом элементарном взаимодействии один активный центр образует молекулу продукта реакции и один новый активный центр. Разветвленная цепная реакция характеризуется тем, что по ходу взаимодействия свободного радикала с молекулой исходного реагента происходит образование нескольких новых активных центров, одни из которых дают начало новым активным центрам, а другие продолжают старую цепь.
Пример разветвленной цепной реакции – реакция образования воды из простых веществ:
Теория разветвленных цепных реакций была выдвинута Н.Н. Семеновым в 20-х годах XX века при изучении кинетики разнообразных процессов. Теория цепных реакций является научной основой для отраслей техники. Ядерные цепные реакции тоже относятся к цепным процессам.
ЦЕПНЫЕ РЕАКЦИИ – химические реакции, идущие путем последовательности одних и тех же элементарных стадий, на каждой из которых возникает одна или несколько активных частиц (атомов, свободных радикалов, ионов, ион-радикалов). По цепному механизму протекают реакции крекинга, горения, полимеризации и ряд других реакций
Цепи Боденштейна – Нернста. К концу 19 в. была разработана важнейшая глава физической химии – учение о равновесиях химических реакций (химическая термодинамика). Стало возможным рассчитывать, на какую максимально возможную глубину может пройти конкретная реакция при данных условиях. Одновременно создавалось учение о скоростях химических процессов – химическая кинетика. Накопленные ко второй половине 19 в. многочисленные экспериментальные данные можно было объяснить на основании закона действующих масс и уравнения Аррениуса. В то же время появлялись факты, которые невозможно было объяснить ни одной из существовавших теорий. Одной из самых загадочных оказалась очень простая с виду реакция водорода с хлором: H2 + Cl2 ® 2HCl.
В 1845 английский химик Джон Дрепер обнаружил, что под действием солнечного света хлор приобретает особую активность в реакции с водородом (см. ФОТОХИМИЯ). Еще более удивительный факт обнаружили в 1857 немецкий химик Роберт Бунзен и его ученик из Англии Генри Роско. Оказалось, что некоторые примеси даже в самых малых концентрациях могут оказать огромное влияние на скорость этой реакции. Например, малые добавки кислорода замедляли ее в сотни раз. Это был парадоксальный результат, так как кислород сам прекрасно реагирует с водородом. Обнаружились и другие непонятные явления. Например, скорость реакции зависела от материала стенки сосуда и даже от его размеров. В стройном, казалось бы, учении о скоростях реакций появилась брешь, и никто не знал, как с ней справиться.
А реакция водорода с хлором преподносила ученым все новые сюрпризы. В начале 20 в. Альберт Эйнштейн сформулировал закон, согласно которому каждый поглощенный квант света (фотон) вызывает изменения лишь в одной молекуле. Экспериментально несложно измерить число прореагировавших (или образовавшихся) молекул и число поглощенных в реакции квантов света. Отношение этих величин называется квантовым выходом реакции. Так, если на каждый поглощенный реагентами квант света образуется одна молекула продукта, то квантовый выход такой реакции равен единице. Однако экспериментально измеренные квантовые выходы многих реакций не соответствовали закону квантовой эквивалентности. В 1913 один из основоположников химической кинетики немецкий химик Макс Боденштейн измерил квантовый выход фотохимической реакции водорода с хлором H2 + Cl2 ® 2HCl. Результат оказался невероятным: число молекул HCl, образовавшихся при поглощении смесью одного кванта света, в некоторых условиях достигал миллиона! Боденштейн объяснил этот поразительный результат единственным разумным методом: каждый поглощенный квант света «запускает» длинную цепочку превращений, в которой реагируют сотни тысяч молекул исходных веществ (H2 и Cl2), превращаясь в молекулы продукта реакции (HCl). Это похоже на то, как выстроенные в ряд костяшки домино быстро, как по команде, падают одна за другой, если удачно толкнуть первую из них.
Боденштейном были сформулированы и основные принципы протекания нового типа химических превращений – цепных реакций. Эти реакции обязательно имеют три стадии: 1) зарождение цепи, когда происходит образование активных частиц; 2) продолжение (развитие) цепи; 3) обрыв цепи. Зарождение цепей в тепловой реакции происходит в результате диссоциации молекул при нагревании. В фотохимической реакции зарождение цепей происходит при поглощении кванта света. На стадии продолжения цепи образуются молекулы продуктов реакции и одновременно появляется новая активная частица, способная продолжать цепь. На стадии обрыва происходит исчезновение (дезактивация) активной частицы.
При сильном нагреве или при интенсивном освещении ультрафиолетовым светом цепная реакция водорода с хлором идет со взрывом. Но если температура не очень высокая или интенсивность света невелика, реакция идет спокойно. Основываясь на этом факте, Боденштейн выдвинул очень важный принцип стационарной концентрации промежуточных продуктов цепных реакций. В соответствии с этим принципом, скорость генерирования активных частиц на стадии зарождения равна скорости их исчезновения на стадии обрыва. Действительно, если бы скорость обрыва была больше скорости зарождения цепей, число активных частиц снизилось бы до нуля, и реакция прекратилась бы сама собой. В случае же преобладания скорости зарождения, число активных частиц росло бы со временем, что привело бы к взрыву.
Однако выяснение химического механизма для каждой стадии реакции водорода с хлором оказалось трудной задачей. Боденштейн предположил теорию энергетического разветвления: образующиеся в первичной реакции молекулы HCl несут избыточную энергию и потому способствуют протеканию дальнейших реакций, передавая избыток энергии молекулам исходных веществ. Однако эта теория в данном случае оказалась неверной. Правильный механизм реакции дал в 1918 немецкий физикохимик лауреат Нобелевской премии Вальтер Нернст. Он предположил, что активными частицами являются атомы водорода и хлора; при этом схема цепной реакции выглядела так. Зарождение цепи происходит при термической диссоциации молекул хлора при высокой температуре или же при поглощении ими квантов света при комнатной температуре: Cl2 ® 2Cl. Далее следуют две быстро повторяющиеся одна за другой стадии продолжения цепи: Cl + H2 ® HCl + H и H + Cl2 ® HCl + Cl. Обрыв цепей происходит, когда активные атомы водорода или хлора реагируют с молекулами примеси, или «прилипают» к стенке сосуда, или реагируют (рекомбинируют) друг с другом, превращаясь в неактивные молекулы H2 и Cl2.
В последующем было показано, что атомы водорода намного активнее атомов хлора; соответственно атомы водорода реагируют намного быстрее и потому их стационарная концентрация значительно ниже. Так, при комнатной температуре стационарная концентрация атомов водорода примерно в 100 раз меньше, чем атомов хлора. В результате вероятность встречи двух атомов водорода или атомов водорода и атомов хлора намного меньше, чем для двух атомов хлора, поэтому практически единственной реакцией обрыва цепей является рекомбинация атомов хлора: Cl + Cl ® Cl2. Если давление в реакционном сосуде очень мало, а его размеры невелики, активные частицы могу достигнуть стенки сосуда еще до того, как прореагируют с молекулами H2 и Cl2; в этих условиях важную роль может приобрести обрыв цепей на стенках реакционного сосуда.
Схема Нернста была подтверждена разными экспериментами. Один из самых остроумных провел английский физикохимик Майкл Поляни. В его опытах струя водорода проходила над слегка подогретым металлическим натрием и уносила с собой некоторое очень небольшое количество его паров. Затем струя попадала в темноте в сосуд с хлором. При температуре опыта чистый водород с хлором не реагировал, но ничтожная примесь паров натрия полностью меняла дело: шла быстрая реакция образования хлороводорода. Здесь роль инициатора цепной реакции вместо света играет натрий: Na + Cl2 ® NaCl + Cl. Подобно тому, как в фотохимической реакции на каждый поглощенный квант света приходится очень много прореагировавших молекул, так и здесь на каждый прореагировавший атом натрия приходится много образовавшихся молекул HCl. Аналогичные результаты Поляни получил для реакции хлора с метаном. В этом случае реакции инициирования и обрыва цепей были такими же, как в реакции хлора с водородом, а реакции продолжения цепи выглядели так: Cl· + CH4 ® HCl + ·CH3 и ·CH3 + Cl2 ® CH4 + Cl·. В этих реакциях также участвуют частицы с неспаренными электронами (обозначены точкой) – свободные радикалы.
Многие реакции с участием свободных радикалов оказались цепными, механизм которых в общих чертах был сходен с механизмом реакции водорода с хлором. По цепному механизму протекают реакции расщепления при высоких температурах (пиролиза) углеводородов, например, этана: C2H6 ® C2H4 + H2; подобные реакции имеют большое значение при промышленной переработке углеводородов нефти. Цепными оказались реакции окисления органических соединений кислородом, реакции присоединения к непредельным соединениям галогенов (хлора и брома), бромоводорода и других соединений, реакции полимеризации, ряд других процессов. Цепные реакции полимеризации интересны тем, что в них стадии продолжения цепи оставляют за собой «реальные цепи» в виде связанных друг с другом остатков мономерных звеньев. В загустевшем и затвердевшем полимере (например, в полистироле или в полиметилметакрилате – «органическом стекле») иногда можно обнаружить концевые свободные радикалы, которым из-за высокой вязкости не удалось прореагировать со свободной молекулой мономера.
Цепи Семенова – Хиншелвуда. В конце 1924 в Ленинградском Физико-техническом институте, в Лаборатории электронной химии, которой заведовал Н.Н.Семенов, начали измерять интенсивность свечения паров фосфора при их окислении кислородом. В первых же опытах молодая выпускница университета Зинаида Вальта и ее непосредственный руководитель, Ю.Б.Харитон натолкнулись на совершенно неожиданное явление. Оказалось, что когда кислорода мало, реакция окисления фосфора вообще не идет. Но стоит давлению кислорода превысить некоторое критическое значение, начиналось интенсивное окисление с испусканием света. До этого теория предполагала, что скорость реакции должна плавно увеличиваться с увеличением концентрации. Здесь же – резкий переход от полного отсутствия реакции к очень быстрому процессу при ничтожном изменении давления. Выяснился еще один, совсем уже странный факт: при давлении ниже критического, т.е. в отсутствие реакции, достаточно было ввести в сосуд аргон, чтобы произошла яркая вспышка. Получалось, что инертный газ аргон, не способный ни к каким химическим реакциям, делал кислород реакционноспособным! Это было уже настоящим чудом...
Позже выяснилось, что кислород может полностью терять свою активность не только при снижении, но и при повышении давления выше некоторого критического значения. Этот второй (верхний) предел давления кислорода существенно зависел от примесей различных веществ. Некоторые из таких примесей делали «пассивный» кислород весьма активным, вызывающим горение фосфора. Такое поведение противоречило всем существовавшим тогда представлениям о механизмах и скоростях химических реакций.
Результаты странных экспериментов, без какой-либо попытки их объяснения, были опубликованы в немецком «Физическом журнале». Последствия были быстры и неутешительны: работа подверглась крайне острой критике со стороны знаменитого Боденштейна, который к тому времени считался главой мировой химической кинетики. Он написал, что все результаты по окислению фосфора являются не открытием, а иллюзией и указал даже на ее причину – неправильную конструкцию установки, в которой проводились опыты. В заключение своей короткой статьи Боденштейн отмечал, что так называемые «предельные» явления неоднократно наблюдались в прошлом для разных реакций, но при проверке каждый раз оказывалось, что все они связаны с различными экспериментальными ошибками.
Возражения были очень серьезны. Но тщательная проверка (уже без Харитона – он был в заграничной командировке и без Вальта – она перешла в другой институт) показала правильность первой публикации. Более того, были получены новые, не менее «еретические» данные. Оказалось, например, что критическое давление кислорода сильно зависит от размеров реакционного сосуда.
Семенов почувствовал, что стоит на пороге открытия. Он понимал, что реакция является цепной, наподобие реакции водорода с хлором. Однако механизм цепной реакции Боденштейна – Нернста, основанный на «принципе домино», никогда не приводил (и не мог приводить) к критическим явлениям. Здесь было что-то иное. Одновременно в этом направлении начал работать в Англии Сирил Хиншелвуд. В обеих лабораториях критические явления были обнаружены в реакциях горения водорода и ряда других веществ. Оказалось, например, что в стеклянных термостойких сосудах при температурах 500–600° С реакция водорода с кислородом не идет вовсе, пока давление не достигнет 3–4 мм рт. ст. Когда давление превышало этот нижний предел, внезапно начиналась быстрая реакция, сопровождающаяся свечением. Но самое поразительное явление заключалось в том, что пламя можно было потушить, просто повысив давление. При температуре же ниже 400° С воспламенение в чистой смеси водорода с кислородом не наблюдалось ни при каких давлениях. Однако достаточно было добавить к смеси инертный газ, как происходила вспышка!
Все эти новые явления были объяснены Семеновым (и независимо Хиншелвудом) в предположении о разветвляющихся цепях. Если в реакции водорода с хлором на каждой стадии продолжения цепи одна активная частица расходуется и одна – появляется (неразветвленная цепь), то в реакции водорода (и других реагентов) с кислородом на одну исчезнувшую активную частицу образуется две или более новых, например, H + O2 ® OH + O O + H2 ® OH + H OH + H2 ® H2O + H
Если сложить эти три последовательные реакции, получим Н + О2 + 2Н2 ® ОН + 2Н, то есть одна активная частица превращается в три. В результате число активных центров стремительно нарастает (цепи разветвляются), и если скорость обрыва цепей недостаточно велика, реакция очень быстро переходит во взрывной режим (при небольшом давлении вместо взрыва наблюдается вспышка). Такие реакции, идущие с увеличение числа активных частиц, назвали разветвленно-цепными. Если учесть, что эти процессы сильно экзотермичны, а для реакции каждой активной частицы с молекулой исходного вещества требуются миллиардные доли секунды, то легко понять, почему разветвленно-цепные реакции при больших концентрациях (давлениях) реагентов вызывают разрушительные взрывы.
Важно отметить, что лавина разветвленно-цепной реакции очень быстро заканчивается: спустя доли секунды после ее начала для продолжения реакции уже не хватает исходных веществ – они почти все превратились в продукты реакции. Здесь можно привести такую аналогию: по «разветвленно-цепному механизму» распространяются различные слухи, если каждый узнавший новость расскажет ее более чем одному человеку. И так же, как слухи и сплетни, быстро распространяются, но и быстро заканчиваются разнообразные «разветвленно-цепные» финансовые и прочие пирамиды (типа знаменитой «Властилины», МММ, «писем счастья» и т.п.), различные «заманчивые» предложения за 100 рублей заработать 100 тысяч и другие аферы, требующие на каждом этапе привлечения новых «клиентов». С первого взгляда все выглядит честно, но в пирамиду быстро вовлекается все большее число участников, и вскоре не хватает «исходных веществ» – некому больше покупать акции, и они стремительно обесцениваются. Подобные финансовые пирамиды еще в 19 в. были в ходу в разных странах; во Франции их называли «снежным комом», у нас – лавиной. Их механизм (и математическое описание) весьма напоминают разветвленно-цепные химические реакции.
Семенов и Хиншелвуд дали такое объяснение изученным процессам. При низких давлениях большинство активных частиц – атомов, свободных радикалов, не успев столкнуться со многими молекулами реагентов и «размножиться», долетают до стенки реакционного сосуда и «погибают» на них – цепи обрываются. Чем меньше диаметр реактора, тем больше у радикалов шансов достичь его стенок. Вот откуда зависимость от размеров сосуда! С повышением концентрации шансов столкнуться с молекулами реагентов для радикалов становится больше, чем шансов достичь стенки – возникает лавина реакций. Это объясняет существование нижнего предела по давлению. Молекулы инертного газа, по меткому выражению Семенова, «путаясь в ногах» у активной частицы, замедляют ее движение к стенке; так объясняется удивительное влияние аргона на величину критического давления.
Когда же достигается верхний предел по давлению, цепи снова обрываются быстрее, чем происходит их разветвление; однако причина обрыва цепей здесь иная – активные радикалы исчезают в результате «взаимного уничтожения» – рекомбинации в объеме сосуда (скорость этой реакции очень быстро увеличивается с ростом давления). Таким образом, все экспериментальные факты получили логичное объяснение в рамках теории разветвленной цепной реакции. В 1956 Н.Н.Семенов и С.Хиншелвуд за эти исследования получили Нобелевскую премию по химии.
Теория разветвленно-цепных реакций имеет большое практическое значение, так как объясняет поведение многих промышленно важных процессов, таких как горение, крекинг нефти, воспламенение горючей смеси в двигателях внутреннего сгорания. Наличие верхнего и нижнего пределов по давлению означает, что смеси кислорода с водородом, метаном, другими горючими газами взрываются лишь при их определенных соотношениях. Например, смеси водорода с воздухом взрываются при содержании водорода от 4 до 75%, а смеси метана с воздухом – при содержании метана от 5 до 15%. Вот почему так опасны утечки газа: если метана в воздухе окажется больше 5%, взрыв может наступить даже от крошечной искры в выключателе при включении или выключении света на кухне.
Особое значение цепные процессы приобрели в связи с работой физиков по получению ядерной энергии. Оказалось, что деление урана, плутония, других расщепляющихся материалов подчиняется тем же закономерностям, что и разветвленно-цепные химические реакции. Так, реакция деления урана вызывается нейтронами, которые расщепляют ядра урана с выделением огромной энергии. Разветвление цепи происходит за счет того, при расщеплении ядра выделяются несколько активных частиц – нейтронов, способных к расщеплению новых ядер.
Реакции с вырожденным разветвлением. При окислении некоторых соединений получаются пероксиды, которые сами способны в определенных условиях распадаться с образованием активных частиц – свободных радикалов. В результате происходит разветвление цепей, хотя и не такое быстрое: ведь чтобы распад пероксидов шел с заметной скоростью, они должны сначала накопиться. Такие процессы назвали вырожденными разветвлениями.
Типичный пример разветвленно-цепной реакции с вырожденным разветвлением – реакция окисления углеводородов. Она начинается с того, что молекула кислорода отрывает от молекулы органического соединения атом водорода: RH + O2 ® R· + HO2·. Гидропероксидный радикал, образовавшийся на стадии инициирования, в результате реакции HO2· + RH ® H2O2 + R· превращается в радикал R· с неспаренным электроном на атоме углерода. Так что радикал HO2· далее в реакции не участвует. У радикала R· несколько возможностей. Во-первых, он может соединяться (рекомбинировать) с другими радикалами, в том числе и себе подобными: R· + R· ® R2. Во-вторых, может отрывать атом водорода от молекулы исходного вещества: R· + R'H ® RH + R'·. Наконец, он может присоединяться по двойной связи молекулы кислорода: R· + O=O ® R–O–O·. Первую реакцию можно не принимать во внимание: вероятность встречи двух активных радикалов очень мала, так как их концентрация ничтожна. Вторая реакция приводит лишь к обмену атомом водорода. А вот в результате третьей реакции образуется пероксидный радикал RO2·, который вместе с радикалом R· ведет цепь. Она состоит из двух повторяющихся стадий цепной реакции окисления: RO2· + RH ® ROOH + R· и R· + O2 ® RO2·.
Видно, что цепь ведут радикалы RO2· и R·, поскольку именно они постоянно рождаются в ходе реакции. Радикалы RO2· менее активные, их концентрация намного выше, поэтому цепь обрывается, когда встречаются два пероксидных радикала. Эта встреча может дать различные продукты, в числе которых пероксиды ROOR (они образуются при рекомбинации пероксидных радикалов), спирты, карбонильные соединения. Если цепи длинные, этих веществ – продуктов рекомбинации – будет немного, а основным продуктом цепной реакции станет гидропероксид ROOH, который иногда удается получить с высоким выходом. Связь О–О в гидропероксидах относительно слабая (более чем вдвое слабее связи С–О в спиртах). При ее разрыве образуются сразу два радикала – RO· и OH·, которые инициируют новые цепи. Получается, что продукт реакции, гидропероксид, одновременно ускоряет ее. Такие реакции называются автокаталитическими.
Возрождение «энергетических цепей». Высказанное Боденштейном и рядом других химиков предположение об «энергетических цепях» не получило экспериментального подтверждения и было на многие десятилетия забыто. Однако в 1963 В.И. Веденеев, А.М. Чайкин и А.Е. Шилов обнаружили, что «энергетические разветвления» возможны в реакциях фторирования ряда соединений. Примером может служить реакция фтора с водородом. В этой реакции на стадии продолжения цепи H + F2 ® HF* + F выделяется так много энергии, что образующаяся «горячая» молекула фтороводорода (она помечена звездочкой) может вызвать разветвление цепи. Происходит это путем передачи избыточной энергии исходным веществам; переносчиком энергии при этом является молекула водорода. Механизм реакции таков: F2 + H2 ® H + HF + F – медленная стадия зарождения цепи F + H2 ® HF + H – две реакции
H + F2 ® HF* + F – продолжения цепи HF* + H2 ® HF + H2* – передача возбуждения
H2* + F2 ® H + HF* + F – разветвление цепи Обрыв цепей происходит на молекулах примесей или на стенках сосуда. Исследование механизма этой реакции позволило создать химический фтор-водородный лазер, в котором источником света (в инфракрасном диапазоне) являются возбужденные молекулы HF.
Комплексные соединения. Строение, номенклатура, применение
Комплексные соединения
Комплексные соединения представляют наиболее распространенную и неоднородную группу химических веществ. К ним относится огромное количество минералов и природных металлоорганических соединений (хлорофилл, гемоглобин, витамин В12 и т.п.), а также множество искусственно синтезированных веществ, которые приобрели важное значение в прикладной химии, химической технологии и почти во всех без исключения областях хозяйства.Учитывая многочисленность комплексных соединений и разнообразие присущих им свойств, не удается сформулировать однозначное определение, которое охватывало бы все разновидности этого класса веществ. Однако инженер в практической деятельности наиболее часто имеет дело с соединениями, для которых справедливой считается такая характеристика.
Комплексными называются соединения, в узлах кристаллических решеток которых размещаются сложные ионы, построенные за счет координации определенных частиц вокруг центрального атома (иона) и способные к самостоятельному существованию при переходе вещества в растворенное или расплавленное состояние. Между комплексными соединениями и некоторыми обычными веществами невозможно провести резкую границу. Более того, одно и то же вещество в зависимости от условий может вести себя поразному. Например, в твердом состоянии роданид состава Pb(CNS)2· 4KCNS содержит в узлах кристаллической решетки сложные ионы [Pb(CNS)6]4-, которые не разрушаются и при его растворении в органических растворителях, способствующих протеканию электролитической диссоциации:
Координационная теория комплексных соединений К концу ХIХ в. был накоплен огромный материал о свойствах комплексных соединений, однако их строение и состав не удавалось объяснить с позиций классической теории валентности.Основатель координационной теории А.Вернер (1893г.) предположил, что в отличие от обычных веществ в комплексных соединениях элементы проявляют, кроме главной валентности, еще и дополни-тельную, побочную. Благодаря действию сил именно побочной валентности происходит процесс комплексообразования. Но в самом комплексе различие между главной и побочной валентностями исчезает, все связи становятся равноценными, их называют координационными.
Вернер подчеркнул, что действие побочной валентности вызывает укрепление связей между атомами. Например, соединение PbCl4 чрезвычайно неустойчиво, а комплексное соединение K2[PbCl6], образование за счет побочной валентности свинца(+2), -- довольно прочное. Таким образом, в комплексных соединениях наблюдается стабилизация как высших, так и низших валентных форм элемента.
В современном виде координационная теория А.Вернера, которая постоянно углубляется и дополняется, сводится к нескольким основным положениям.
1. В молекуле комплексного соединения атом (ион), который за счет главной и побочной валентностей координирует вокруг себя определенное количество нейтральных молекул или противоположно заряженных ионов, называется центральным атомом, или комплексообразователем.
Наиболее часто в роли комплексообразователя выступают положительно заряженные ионы (реже -- атомы) d- и р-металлов, иногда -- щелочноземельных и даже щелочных металлов, а также некоторых неметаллов ( В, Si, P, As).
2. С центральным атомом непосредственно соединяются молекулы или ионы, которые называются координированными группами, аддендами, или лигандами.
3. Комплексообразователь совместно с лигандами составляет внутреннюю сферу комплексного соединения, или просто комплекс, который при написании координационной формулы берется в квадратные скобки, чтобы подчеркнуть его монолитность: К3[Fe(CN)6], Na[BF4].
Заряд внутренней сферы определяется алгебраической суммой степени окисления комплексообразователя и совместных зарядов всех лигандов:
4. Если степень окисления комплексообразователя по абсолютной величине не равна сумме зарядов всех лигандов, то комплексное соединение содержит внешнюю сферу, которую записывают вне квадратных скобок: [Ag(CN)2]Cl, K[Ag(NH3)2]. Однако существуют и нейтральные комплексы, лишенные внешней сферы, их иногда называют комплексными неэлектролитами:
5. Общее количество координационных валентностей, с помощью которых комплексообразователь во внутренней сфере связан с лигандами, называется координационным числом (к.ч.).
Известны координационные числа от 1 до 9 и 12. Наиболее распространенными являются комплексные соединения с координационными валентностями 2, 4 и 6. Довольно часто координационное число бывает вдвое большим, чем степень окисления комплексообразователя, например:
однако это нестрогая зависимость.
В общем случае координационное число может иметь переменную величину не только для разных комплексообразователей, но и для одного и того же, поскольку на количество координированных лигандов влияют определенные факторы:
1. Заряд лиганда. Например, координационное число никеля (+2) с нейтральными лигандами равно 6, а с отрицательно заряженными анионами CN-, между которыми действуют значительные силы отталкивания, -- только 4:
2. Размеры лигандов. Комплексы, содержащие небольшие по размерам ионы, характеризуются большими величинами координационных чисел, например, для фторсодержащих алюминатных комплексов координационное число больше, чем для хлор- и бромсодержащих, так как радиус фторид-иона намного меньше, чем радиусы ионов Cl- и Br-.
Свойства растворителя, в котором происходит образование комплексного соединения. Так, в полярном растворителе -- воде -- легче возникают комплексы [Co(CNS)1]+ и [Co(CNS)2], а в малополярном -- ацетоне -- при одних и тех же условиях -- [Co(CNS)3]- и [Co(CNS)4]2-
· Концентрация реагирующих компонентов. При возрастании концентрации появляется тенденция к образованию более сложных комплексов. Например, в разбавленных растворах удается идентифицировать лишь комплекс [Fe(CNS)1]2+, в то время, как с увеличением концентраций реагирующих веществ получаются более сложные комплексы: от [Fe(CNS)3] вплоть до [Fe(CNS)6]3-
Энергетические эффекты в химических реакциях. Внутренняя энергия. Энтальпия. Стандартная энтальпия образования химических соединений
Энергетические эффекты химических реакций изучает термохимия. Данные об энергетических эффектах используются для выяснения направленности химических процессов, для расчета энергетических балансов технологических процессов и т.д. С их помощью можно рассчитать температуру горения различных веществ и материалов, температуру пожаров и т.п.
Состояние системы (вещества или совокупности рассматриваемых веществ) описывают с помощью ряда параметров состояния – t, p, m. Для характеристики состояния системы и происходящих в ней изменений важно знать также изменение таких свойств системы, как ее внутренняя энергия U, энтальпия Н, энтропия S, энергия Гиббса (изобарно-изотермический потенциал) G. По изменению этих свойств системы можно судить, в частности, об энергетике процессов.
Химические реакции обычно протекают при постоянном объеме V = const, DV = 0 (например, в автоклаве) или при постоянном давлении p = const (например, в открытой колбе), т.е. является соответственно изохорными или изобарными процессами.
Энергетический эффект химического процесса возникает за счет изменения в системе внутренней энергии U или энтальпии H. Внутренней энергией системы называют энергию всех видов движения и взаимодействия тел или частиц, составляющих систему (кинетическая энергия межмолекулярного взаимодействия, вращательная энергия, колебательное движение атомов и групп в молекуле, энергия взаимодействия электронов между собой и с ядрами).
Обычно химические реакции сопровождаются тепловыми эффектами. Тепловым эффектом называется суммарное количество энергии, выделенной или поглощенной системой в результате реакции, проводимой при постоянной температуре. Раздел химии, который изучает тепловые эффекты химических реакций и фазовых превращений, называется термохимией.
Согласно первому закону термодинамики (уравнение 4.6) количество выделенной или поглощенной системой теплоты Q определяется равенством:
Q = DU + W.
Подставив выражение (4.5) в (4.6), получим равенство:
Q = DU + p·DV, (4.7).
определяющее тепловой эффект химической реакции. Из равенства (4.7) следует, что тепловой эффект реакции зависит от того, в каких условиях она протекает. В изохорном процессе V = const, DV = 0, следовательно, тепловой эффект реакции QV равен изменению внутренней энергии системы:QV = U2 – U1 = DU, т.к. W = 0 (4.8).
В изобарном процессе p = const, следовательно, тепловой эффект реакции Q равен:
QP = DU + p·DV = (U2 – U1) + p·(V2 – V1) = (U2 + p·V1) - (U1 + p·V1).
Обозначим:U + p·DV = H (4.9).
Величина H называется энтальпией или теплосодержанием системы. Поэтому тепловой эффект химической реакции при изобарном процессе равен изменению энтальпии системы:
QP = H2 – H1 = DH (4.10).
или
QP = DU + p·DV = DH (4.10а).
Энтальпия, также как и внутренняя энергия, является термодинамической функцией состояния системы.
Для реакций, в которых участвуют только твердые и жидкие вещества, член p·DV в уравнении (4.10а) пренебрежимо мал или равен нулю. Для подобных реакций выполняется соотношение DH » DU. Для газофазных реакций, протекающих с участием газообразных веществ, изменение объема значительно. Если DV > 0, т.е. происходит расширение, то DH > DU; если DV < 0, т.е. происходит сжатие, то DH < DU. Произведение p·DV для таких реакций можно рассчитать из уравнения идеального газа:p·DV = n·R·T или
p·DV = Dn·R·T, где Dn - изменение числа моль газа, определяемое из уравнения реакции; например,
(NH4)2Cr2O7 = Cr2O3 + N2 + 4H2O, Dn = 5.
Химические реакции, протекающие с выделением теплоты, называются экзотермическими. При этом в изохорном процессе внутренняя энергия системы уменьшается, т.е. DU < 0 (т.к. U2 < U1), а в изобарном процессе - энтальпия уменьшается, т.е. DH < 0 (т.к. H2 < H1) (рис.4.2).
Химические реакции, протекающие с поглощением теплоты, называются эндотермическими. При этом в изохорном процессе DU > 0, в изобарном процессе - DH > 0. Уменьшение энтальпии в экзотермических процессах означает, что суммарная энергия, содержащаяся в продуктах реакции в виде энергии химических связей, межмолекулярных взаимодействий, молекулярных колебаний и т.д. меньше суммарной энергии исходных веществ (реагентов). И наоборот, увеличение энтальпии в эндотермических процессах означает, что суммарная энергия, содержащаяся в продуктах реакции больше суммарной энергии исходных веществ.
Изменение энтальпии при стандартном состоянии веществ, участвующих в реакции или при фазовом превращении, обозначается DH°(T) и DH°(298 K), если температура системы T или 298,15 K.
Тепловые эффекты химических реакций зависят не только от условий (температура, давление, объем), в которых они протекают, но и от количества веществ, участвующих в реакции, и их физического состояния. Поэтому для того, чтобы можно было сравнивать энергетические эффекты различных процессов, их характеризуют изменением энтальпии при стандартных условиях, соответствующим конкретному уравнению химической реакции. Уравнения химических реакций, в которых указаны их тепловые эффекты и агрегатные состояния (г-газовое, ж-жидкое, к-кристаллическое, т-твердое) или аллотропные модификации (например, a-сера, b-сера) веществ, называются термохимическими уравнениями реакций. Например:
2H2(г) + O2(г) = 2H2O(ж), DH°(298 K) = -571,6 кДж 2H2(г) + O2(г) = 2H2O(г), DH°(298 K) = -483,6 кДж
ТЕРМОХИМИЧЕСКИЕ ЗАКОНЫ. ТЕРМОХИМИЧЕСКИЕ РАСЧЕТЫ
Тепловые эффекты химических реакций можно определить экспериментально или расчетным путем. Измерение тепловых эффектов называется калориметрией. В основе термохимических расчетов лежит закон, сформулированный русским ученым Г.И. Гессом (1840 г.):
Тепловой эффект химической реакции не зависит от пути ее протекания, а зависит от природы и физического состояния исходных веществ и продуктов реакции.Это означает, что если какую-либо реакцию представить в виде нескольких последовательных стадий, то тепловой эффект данной реакции будет равен сумме тепловых эффектов каждой стадии.
Например, тепловой эффект реакции горения метана равен DH° = -890,2 кДж
(1) CH4 + 2O2 = CO2 + 2H2O ; DH°1 = -890,2 кДжПусть это превращение представляет собой “Путь А”, проходящий через стадию (1). Можно представить протекание данной реакции через “Путь В”, проходящий через ряд промежуточных стадий (2), (3), (4) и (5), где стадия (5) = (3) + (4). Тепловые эффекты каждой из этих стадий равны соответственно:
(2) CH4(г) = C(графит) + 2H2(г) ; DH°2 = +74,9 кДж
(3) C(графит) + O2(г) = CO2(г) ; DH°3 = -393,5 кДж
(4) 2H2(г) + O2(г) = 2H2O(ж) ; DH°4 = -571,6 кДж
Согласно закону Гесса сумма тепловых эффектов на каждой стадии “Пути В” будет равна тепловому эффекту реакции горения метана на “Пути А”:
DH°1 = DH°2 + DH°5= DH°2 + DH°3 + DH°4 -890,2 = 74,9 - 393,5 - 571,6 (кДж)
Экспериментально было установлено (закон Ломоносова - Лавуазье - Лапласа), что тепловые эффекты прямой и обратной реакций численно равны, но противоположны по знаку.
Так, если прямая реакция экзотермическая, то обратная - эндотермическая:
Из закона Гесса вытекают два важных в практическом отношении следствия.
Первое следствие из закона Гесса: тепловой эффект реакции равен сумме энтальпий (теплот) образования продуктов реакции за вычетом суммы энтальпий (теплот) образования исходных веществ с учетом стехиометрических коэффициентов в уравнении реакции.
Так для реакции, протекающей по уравнению:
aA + bB = pP + qQ,
тепловой эффект рассчитывается по формуле:
DH = [pDfH(P) + qDfH(Q)] - [aDfH(A) + bDfH(B)] (4.11).
Энтальпия (теплота) образования - это тепловой эффект реакции образования 1 моль сложного вещества из простых веществ: DfH [Дж/моль; кДж/моль]. Обычно в расчетах используют стандартные энтальпии образования. Стандартная энтальпия образования DfH°(298 K) это тепловой эффект образования 1 моль сложного вещества из простых веществ при стандартных условиях (T = 298,15 K и p = 101,3 кПа). Стандартные энтальпии образования простых веществ, устойчивых в стандартных условиях (газообразный O2, кристаллический I2 и т.д.) принимают равными нулю. Например, окисление водорода можно представить тремя уравнениями:
(6) 2H2(г) + O2(г) = 2H2O(ж) ; DH°(298K)6 = -571,6 кДж
(7) H2(г) + 1/2O2(г) = H2O(ж) ; DH°(298K)7 = -285,8 кДж
(8) H2(г) + 1/2O2(г) = H2O(г) ; DH°(298K)8= -241,8 кДж
Каждому уравнению соответствует определенное значение теплового эффекта. И только тепловой эффект реакции, описываемой уравнением (7), будет равен стандартной теплоте образования воды DH°(298 K)7 = DfH°(298 K, H2O(ж)). Согласно этому уравнению в реакции образуется 1 моль воды, стандартным состоянием которой при 298 K является жидкое.Для многих веществ стандартные теплоты образования известны и сведены в справочные таблицы.Теплота образования является мерой термодинамической устойчивости (прочности) сложного вещества относительно простых веществ, из которых оно образовано. Можно утверждать, что чем более отрицательное значение имеет стандартная энтальпия образования вещества, тем оно устойчивее. Согласно закону Ломоносова - Лавуазье - Лапласа теплота (энтальпия) образования сложного вещества равна по величине, но противоположна по знаку теплоте (энтальпии) разложения вещества (DdH°(Т)):DfH°(298 K) = |-DdH°(298 K)| Второе следствие из закона Гесса: тепловой эффект реакции равен сумме энтальпий (теплот) сгорания исходных веществ за вычетом суммы энтальпий (теплот) сгорания продуктов реакции с учетом стехиометрических коэффициентов в уравнении реакции.
Так для реакции, протекающей по уравнению: aA + bB = pP + qQ тепловой эффект можно рассчитать по уравнению:DH = [aDсH(A) + bDсH(B)] - [pDсH(P) + qDсH(Q)] Энтальпия (теплота) сгорания - это тепловой эффект сгорания 1 моль горючего вещества до продуктов предельного окисления (до образования высших оксидов): DcH [Дж/моль, кДж/моль].Стандартная энтальпия сгорания DcH°(298 K) - это тепловой эффект реакции сгорания в кислороде 1 моль данного вещества при стандартных условиях. Например, тепловой эффект реакции сгорания 1 моль метана в стандартных условиях равен стандартной теплоте сгорания метана:CH4(г) + 2O2(г) = CO2(г) + 2H2O(г) ; DH°(298 K) = -890,2 кДж, т.е. DH°(298 K) = DcH(298 K, CH4(г)) = -890,2 кДж/моль.Энтальпии сгорания кислорода и высших оксидов равны нулю. Следует четко разграничивать два различных понятия - энтальпия образования и энтальпия сгорания вещества, хотя численные значения этих величин в некоторых случаях совпадают (табл. 4.1). Например, стандартные энтальпии сгорания водорода и углерода равны стандартным энтальпиям образования CO2 и H2O.
Закон Гесса распространяется не только на химические реакции, но и на различные физико-химические процессы, сопровождающиеся энергетическими эффектами: растворение, сольватацию (гидратацию), фазовые превращения (плавление, испарение, возгонка, затвердевание (кристаллизация), конденсация, сублимация)Закон Гесса и его следствия справедливы и используются также для расчета энергии химической связи, энергии кристаллической решетки, энергии межмолекулярного взаимодействия, энтальпии растворения и сольватации (гидратации) и т.д. Закон Гесса справедлив для тех взаимодействий, которые протекают при постоянном объеме или при постоянном давлении, а единственным видом совершаемой работы является работа против сил внешнего давления. Тепловые эффекты реакций, в результате которых совершается другая работа, например, электрическая работа, не могут быть вычислены по закону Гесса, так как их теплоты являются функциями пути.
В каждом веществе запасено определенное количество энергии. С этим свойством веществ мы сталкиваемся уже за завтраком, обедом или ужином, так как продукты питания позволяют нашему организму использовать энергию самых разнообразных химических соединений, содержащихся в пище. В организме эта энергия преобразуется в движение, работу, идет на поддержание постоянной (и довольно высокой!) температуры тела.
Энергия химических соединений сосредоточена главным образом в химических связях. Чтобы разрушить связь между двумя атомами, требуется ЗАТРАТИТЬ ЭНЕРГИЮ. Когда химическая связь образуется, энергия ВЫДЕЛЯЕТСЯ.
Вспомним, что атомы не соединялись бы между собой, если бы это не вело к "выигрышу" (то есть высвобождению) энергии. Этот выигрыш может быть большим или малым, но он обязательно есть при образовании молекул из атомов.
Любая химическая реакция заключается в разрыве одних химических связей и образовании других.
Когда в результате химической реакции при образовании новых связей выделяется энергии БОЛЬШЕ, чем потребовалось для разрушения "старых" связей в исходных веществах, то избыток энергии высвобождается в виде тепла. Примером могут служить реакции горения. Например, природный газ (метан CH4) сгорает в кислороде воздуха с выделением большого количества теплоты (рис. 1-1а). Такие реакции называются ЭКЗОТЕРМИЧЕСКИМИ от латинского "экзо" - наружу (имея в виду выделяющуюся энергию).
В других случаях на разрушение связей в исходных веществах требуется энергии больше, чем может выделиться при образовании новых связей. Такие реакции происходят только при подводе энергии извне и называются ЭНДОТЕРМИЧЕСКИМИ (от латинского "эндо" - внутрь). Примером является образование оксида углерода (II) CO и водорода H2 из угля и воды, которое происходит только при нагревании
Термохимия. Закон Гесса. Следствия из закона Гесса. Теплотворная способность топлив
Раздел химии, посвящённый изучению тепловых эффектов реакций, получил название термохимия.
Термохимия базируется на 2-х основных законах: Если при образовании какого-либо соединения выделяется (или поглощается) некоторое количество теплоты, то при разложении этого соединения в тех же самых условиях такое же количество теплоты поглощается (или выделяется). Результаты термохимических изменений - тепловые эффекты реакций, относят к одному молю образующегося вещества, а количество теплоты, которое выделится при образовании одного моля соединения из простых веществ, называют теплотой образования данного соединения. Если элемент может существовать в виде нескольких простых веществ или алотропных форм, то при расчете теплоты образования этот элемент берут в виде того вещества, которое при данных условиях наиболее устойчиво. Теплообразование наиболее устойчивых при данных условиях простых веществ принимают равным нулю. Теплотообразование менее устойчивых простых веществ равны теплотам их образования из наиболее устойчивых. Химические уравнения, в которых указано количеств выделяющейся, или поглощаемой теплоты, а также агрегатные состояния реагирующих веществ, называют термохимическими уравнениями. Величина теплового эффекта зависит от природы реагирующих веществ, продуктов реакции, их агрегатного состояния и температуры. Для удобства сравнения различных реакций по величинам их тепловых эффектов последние, как правило, указываются для температур исходных веществ, равной 25 ˚С или 298 К, называемой стандартной температурой.
Важнейшей характеристикой веществ, применяемых в качестве топлива служит теплота их сгорания, которая характеризует количество теплоты, выделяющейся при сгорании 1 моля вещества.
Закон Гесса: тепловые эффекты реакций зависят только от начального и конечного состояний веществ и не зависят от промежуточных стадий процесса. Следствие из закона Гесса: тепловой эффект химической реакции равен суме теплот образования получившихся веществ за вычетом суммы теплот образования исходных веществ. Полу-суммирование осуществляется с учётом числа молей, участвующих в реакции веществ в соответствии с её уравнением. Теплотворная способность топлива Качество топлива характеризуется его теплотворной способностью Q и измеряется количеством тепла, выде¬ляющегося при полном сгорании 1 кг твердого или жидкого или I нормального кубического метра (1 нм3)1 газообразного топ¬лива. Для твердого и жидкого топлива эта величина имеет размерность ккал1кг, для газообразного — ккал/нм3.Теплотворная способность рабочей массы топлива обозна¬чается: <?р ккал/кг — для твердого и жидкого топлива и Qp ккал1нм3 — для газообразного.Принято считать, что если теплотворная способность топли¬ва учитывает, кроме тепла, выделившегося при его сгорании, также и тепло, полученное при преобразовании продуктов го¬рения в воду при 100°С, то такая теплотворность называется высшей (QP).
Если при определении теплотворной способности топлива было учтено тепло, расходованное на испарение влаги, такая теплотворность называется низшей (Qp). При практических расчетах пользуются последней.
Теплотворная способность твердого и жидкого топлива определяется либо опытным путем, либо расчетным по формуле Д. И. Менделеева:
Qp = 81Ср + 300НР — 26(0? — SP) ККАЛ/КГ;
Qp = 81Ср + 300НР — 26(Ор — Sp) — 6(9Нр +WP) ККАЛ/КГ.
Теплотворная способность газообразного топлива определя¬ется по формулам, полученным методом суммирования произ¬ведений тепловых эффектов горения газов на их процентное содержание:
QB = 30.5СО + 30,5Н2 + 95,ЗСН4 + 152,5С2Н4 + 4- 60,0H2S ККАЛ/НМ3;
Q„ = 30.5СО + 25,8Н2 + 85,9СН4 + 143,0С2Н4 + 4- 55,2H2S ККАЛ/НМ3.
Для сравнения тепловой ценности различных видов топлива применяется общая единица измерения—так называемое услов¬нее топливо, теплотворная способность которого 7000 ккал/кг. Для перевода любого топлива в условное необходимо действи¬тельную теплотворную способность разделить на 7000 ккал/кг. Полученное отношение называется калорийным эк¬вивалентом. Естественно, что чем больше калорийный эк¬вивалент топлива, тем оно ценнее.Теплотворная способность и калорийные эквиваленты не¬которых наиболее распространенных видов топлива приведены в табл. у. Сжигание топлива и использование тепла отходящих газовТеплотворная способность и калорийные эквиваленты некоторых видов топлива
Горение топлива начинается только в том случав,- когда тем¬пература в топливосжигающем устройстве доведена до темпе¬ратуры воспламенения. Последняя величина переменная и зависит от вида топлива и условий, в которых совершается процесс горения.Горение топлива считается полным, если в продуктах горе¬ния произошло окисление горючих составляющих до ССЬ, ШО, SO2 и -при этом отсутствует свободный кислород.Зная состав топлива, можно подсчитать по формулам, при¬веденным в специальной литературе, теоретическое количество воздуха, необходимое для полного его сжигания. Так как прак¬тически нельзя достичь равномерного смешения воздуха с го¬рючими составляющими топлива, то для обеспечения полного его сгорания необходимо подвести большее количество возудха по сравнению с теоретически необходимым.Отношение количества практически подведенного воздуха 1пр к количеству теоретически необходимому LTeop называется коэффициентом" избытка воздуха:
Величина коэффициента избытка воздуха колеблется в пре¬делах от 1,03 до 1,7. Нижний предел относится к газообразно¬му и жидкому топливу, верхний — к твердому топливу.
Следует помнить, что увеличение избытка воздуха вызывает дополнительный расход тепла для его нагрева, а также увели¬чение количества отходящих продуктов горения, которые уносят большое количество тепла в атмосферу и тем самым снижают температуру нагревательной среды печи. Поэтому необходимо всегда стремиться к полному сжиганию топлива при минималь¬ном коэффициенте избытка воздуха.
Критерием полноты горения служит цвет пламени. При не¬достатке воздуха пламя длинное, красного цвета с черными прожилками. Большой недостаток воздуха вызывает неполное сгорание топлива и появление черного дыма.При избытке воздуха получается короткое, острое (колючее) пламя с ярко светящимися язычками. Такое пламя нагревает маталл неравномерно, вызывает местный его перегрев и даже оплавление. При этом увеличивается расход топлива, угар ме¬талла и вместе с тем снижается температура в печи.
Пламя молочно-белого цвета, без ярких языков в топливо-сжигающем устройстве является признаком оптимального количества воздуха.
Энтропия и ее изменение при химических процессах. Вычисление изменения энтропии.
Энтропия (от греч. entropнa — поворот, превращение), понятие, впервые введенное в термодинамике для определения меры необратимого рассеяния энергии. Э. широко применяется и в других областях науки: в статистической физике как мера вероятности осуществления какого-либо макроскопического состояния; в теории информации как мера неопределенности какого-либо опыта (испытания), который может иметь разные исходы. Эти трактовки Э. имеют глубокую внутреннюю связь. Например, на основе представлений об информационной Э. можно вывести все важнейшие положения статистической физики.В термодинамике понятие «Э.» было введено Р. Клаузиусом (1865), который показал, что процесс превращения теплоты в работу следует общей физической закономерности — второму началу термодинамики. Его можно сформулировать строго математически, если ввести особую функцию состояния — Э.Так, для термодинамической системы, совершающей квазистатически (бесконечно медленно) циклический процесс, в котором система последовательно получает малые количества теплоты dQ при соответствующих значениях абсолютной температуры Т, интеграл от «приведенного» количества теплоты dQ/ Т по всему циклу равен нулю(, т. н. равенство Клаузиуса).
Это равенство, эквивалентное второму началу термодинамики для равновесных процессов, Клаузиус получил, рассматривая произвольный циклический процесс как сумму очень большого, в пределе бесконечного, числа элементарных обратимых Карно циклов. Математически равенство Клаузиуса необходимо и достаточно для того, чтобы выражение
dS = dQ/T (1)
представляло собой полный дифференциал функции состояния S, названное «Э.» (дифференциальное определение Э.). Разность Э. системы в двух произвольных состояниях А и В (заданных, например, значениями температур и объемов) равна (2)
(интегральное определение Э.). Интегрирование здесь ведется вдоль пути любого квазистатического процесса, связывающего состояния А и В, при этом, согласно равенству Клаузиуса, приращение Э. DS = SB — SA не зависит от пути интегрирования.Т. о., из второго начала термодинамики следует, что существует однозначная функция состояния S, которая при квазистатических адиабатных процессах (dQ = 0) остаётся постоянной. Процессы, в которых Э. остаётся постоянной, называются изоэнтропийными. Примером может служить процесс, широко используемый для получения низких температур, — адиабатное размагничивание (см. Магнитное охлаждение). При изотермических процессах изменение Э. равно отношению сообщенной системе теплоты к абсолютной температуре. Например, изменение Э. при испарении жидкости равно отношению теплоты испарения к температуре испарения при условии равновесия жидкости с её насыщенным паром. Согласно первому началу термодинамики (закону сохранения энергии), dQ = dU+pdV, т. е. сообщаемое системе количество теплоты равно сумме приращения внутренней энергии dU и совершаемой системой работы pdV, где р — давление, V — объём системы. С учётом первого начала термодинамики дифференциальное определение Э. принимает вид, (3) откуда следует, что при выборе в качестве независимых переменных внутренней энергии U и объёма V частные производные Э. связаны с абсолютной температурой и давлением соотношениями: Эти выражения представляют собой уравнения состояния системы (первое — калорическое, второе — термическое). Уравнение (4) лежит в основе определения абсолютной температуры (см. также Температура, Температурные шкалы). Формула (2) определяет Э. лишь с точностью до аддитивной постоянной (т. е. оставляет начало отсчёта Э. произвольным). Абсолютное значение Э. позволяет установить третье начало термодинамики, или Нернста теорему: при стремлении абсолютной температуры к нулю разность DS для любого вещества стремится к нулю независимо от внешних параметров. Поэтому: Э. всех веществ при абсолютном нуле температуры можно принять равной нулю (эту формулировку теоремы Нернста предложил в 1911 М. Планк). Основываясь на ней, за начальную точку отсчёта Э. принимают So = 0 при Т = 0. Важность понятия Э. для анализа необратимых (неравновесных) процессов: также была показана впервые Клаузиусом. Для необратимых процессов интеграл от приведённой теплоты dQ / Т по замкнутому пути всегда отрицателен (, т. н. неравенство Клаузиуса). Это неравенство — следствие теоремы Карно: кпд частично или полностью необратимого циклического процесса всегда меньше, чем кпд обратимого цикла. Из неравенства Клаузиуса вытекает, что поэтому Э. адиабатически изолированной системы при необратимых процессах может только возрастать. Т. о., Э. определяет характер процессов в адиабатической системе: возможны только такие процессы, при которых Э. либо остаётся неизменной (обратимые процессы), либо возрастает (необратимые процессы). При этом не обязательно, чтобы возрастала Э. каждого из тел, участвующего в процессе. Увеличивается общая: сумма Э. тел, в которых процесс вызвал изменения. Термодинамическому равновесию адиабатической системы соответствует состояние с максимумом Э. Энтропия может иметь не один, а несколько максимумов, при этом система будет иметь несколько состояний равновесия. Равновесие, которому соответствует наибольший максимум Э., называется абсолютно устойчивым (стабильным). Из условия максимальности Э. адиабатические системы в состоянии равновесия вытекает важное следствие: температура всех частей системы в состоянии равновесия одинакова. Понятие «Э.» применимо и к термодинамически неравновесным состояниям, если отклонения от термодинамического равновесия невелики и можно ввести представление о локальном термодинамическом равновесии в малых, но ещё макроскопических объёмах. Такие состояния можно охарактеризовать термодинамическими параметрами (температурой, давлением и т. д.), слабо зависящими от пространственных координат и времени, а Э. термодинамически неравновесного состояния определить как Э. равновесного состояния, характеризующегося теми же значениями параметров. В целом Э. неравновесной системы равна сумме Э. её частей, находящихся в локальном равновесии. Термодинамика неравновесных процессов позволяет более детально, чем классическая термодинамика, исследовать процесс возрастания Э. и вычислить количество Э., образующейся в единице объёма в единицу времени вследствие отклонения системы от термодинамического равновесия — производство энтропии. Производство Э. всегда положительно и математически выражается квадратичной формой от градиентов термодинамических параметров (температуры, гидродинамической скорости или концентраций компонентов смеси) с коэффициентами, называемыми кинетическими (см. Онсагера теорема).Статистическая физика связывает Э. с вероятностью осуществления данного макроскопического состояния системы. Э. определяется через логарифм статистического веса W данного равновесного состояния S= k ln W (E, N), (7) где k — Больцмана постоянная, W (E, N) — число квантовомеханических уровней в узком интервале энергии DЕ вблизи значения энергии Е системы из N частиц. Впервые связь Э. с вероятностью состояния системы была установлена Л. Больцманом в 1872: возрастание Э. системы обусловлено её переходом из менее вероятного состояния в более вероятное. Иными словами, эволюция замкнутой системы осуществляется в направлении наиболее вероятного распределения энергии по отдельным подсистемам. В отличие от термодинамики статистическая физика рассматривает особый класс процессов — флуктуации, при которых система переходит из более вероятного состояния в менее вероятное, и её Э. уменьшается. Наличие флуктуаций показывает, что закон возрастания Э. выполняется только в среднем для достаточно большого промежутка времеЭ. в статистической физике тесно связана с информационной Э., которая служит мерой неопределённости сообщений данного источника (сообщения описываются множеством величин х1, x2,..., xn, которые могут быть, например, словами какого-либо языка, и соответствующих вероятностей p1, p2,..., pn появления величин x1, x2,..., xn в сообщении). Для определённого (дискретного) статистического распределения вероятностей рк информационной Э. называют величину при условии Значение Ни равно нулю, если какое-либо из pk равно 1, а остальные — нулю, т. е. неопределённость в информации отсутствует. Э. принимает наибольшее значение, когда pk равны между собой и неопределённость в информации максимальна. Информационная Э., как и термодинамическая, обладает свойством аддитивности (Э. нескольких сообщений равна сумме Э. отдельных сообщений). К. Э. Шеннон показал, что Э. источника информации определяет критическое значение скорости «помехоустойчивой» передачи информации по конкретному каналу связи (см. Шеннона теорема). Из вероятностной трактовки информационной Э. могут быть выведены основные распределения статистической физики: каноническое Гиббса распределение, которое соответствует максимальному значению информационной Э. при заданной средней энергии, и большое каноническое распределение Гиббса — при заданных средней энергии и числа частиц в системе. Понятие Э., как показал впервые Э. Шрёдингер (1944), существенно и для понимания явлений жизни. Живой организм с точки зрения протекающих в нём физико-химических процессов можно рассматривать как сложную открытую систему, находящуюся в неравновесном, но стационарном состоянии. Для организмов характерна сбалансированность процессов, ведущих к росту Э., и процессов обмена, уменьшающих её. Однако жизнь не сводится к простой совокупности физико-химических процессов, ей свойственны сложные процессы саморегулирования. Поэтому с помощью понятия Э. нельзя охарактеризовать жизнедеятельность организмов в целом. Д.Н. Зубарев. Э., характеризуя вероятность осуществления данного состояния системы, согласно (7) является мерой его неупорядоченности. Изменение Э. DS обусловлено как изменением р, V и Т, так и процессами, протекающими при р, Т = const и связанными с превращением веществ, включая изменение их агрегатного состояния, растворение и химическое взаимодействие. Изотермическое сжатие вещества приводит к уменьшению, а изотермическое расширение и нагревание — к увеличению его Э., что соответствует уравнениям, вытекающим из первого и второго начал термодинамики (см. Термодинамика): Формулу (11) применяют для практического определения абсолютного значения Э. при температуре Т, используя постулат Планка и значения теплоёмкости С, теплот и температур фазовых переходов в интервале от 0 до Т К. В соответствии с (1) Э. измеряется в кал/(моль К) (энтропийная единица — э. е.) и дж/(моль·К). При расчётах обычно применяют значения Э. в стандартном состоянии, чаще всего при 298,15 К (25 °С), т. е. S0298; таковы приводимые ниже в статье значения Э.
Э. увеличивается при переходе вещества в состояние с большей энергией. D S сублимации > DS парообразования >> DS плавления >DS полиморфного превращения. Например, Э. воды в кристаллическом состоянии равна 11,5, в жидком — 16,75, в газообразном — 45,11 э. е.
Чем выше твёрдость вещества, тем меньше его Э.; так, Э. алмаза (0,57 э. е.) вдвое меньше Э. графита (1,37 э. е.). Карбиды, бориды и другие очень твёрдые вещества характеризуются небольшой Э. Э. аморфного тела несколько больше Э. кристаллического. Возрастание степени дисперсности системы также приводит к некоторому увеличению её Э. Э. возрастает по мере усложнения молекулы вещества; так, для газов N2О, N2O3 и N2O5 Э. составляет соответственно 52,6; 73,4 и 85,0 э. е. При одной и той же молекулярной массе Э. разветвленных углеводородов меньше Э. неразветвлённых; Э. циклоалкана (циклана) меньше Э. соответствующего ему алкена. Э. простых веществ и соединений (например, хлоридов ACIn), а также её изменения при плавлении и парообразовании являются периодическими функциями порядкового номера соответствующего элемента. Периодичность изменения Э. для сходных химических реакций типа 1/n Акрист + 1/2Сl2газ = 1/n ACln крист практически не проявляется. В совокупности веществ-аналогов, например АСl4газ (А — С, Si, Ge, Sn, Pb) Э. изменяется закономерно. Сходство веществ (N2 и СО; CdCl2 и ZnCl2; Ag2Se и Ag2Te; ВаСОз и BaSiO3; PbWO4 и РЬМоО4) проявляется в близости их Э. Выявление закономерности изменения Э. в рядах подобных веществ, обусловленного различиями в их строении и составе, позволило разработать методы приближённого расчёта Э. Знак изменения Э. при химической реакции DS х. р. определяется знаком изменения объёма системы DV х. р.; однако возможны процессы (изомеризация, циклизация), в которых DS х. р. № 0, хотя DV х. р. » 0. В соответствии с уравнением DG = DН — ТDS (G — гиббсова энергия, Н — энтальпия) знак и абсолютное значение DS х. р. важны для суждения о влиянии температуры на равновесие химическое. Возможны самопроизвольные экзотермические. процессы (DG < 0, DH < 0), протекающие с уменьшением Э. (DS < 0). Такие процессы распространены, в частности, при растворении (например, комплексообразование), что свидетельствует о важности химических взаимодействий между участвующими в них веществами.
Энергия Гиббса и ее изменение при химических процессах. Условия самопроизвольного протекания химических реакций
Организм совершает работу, затрачивая внутреннюю энергию, запасенную в виде энергии химического взаимодействия атомов составляющих его веществ. Математическое выражение –ДE = –Q – W первого начала термодинамики определяет точное соотношение между расходом внутренней энергии системы ДЕ, работой W, совершаемой системой, и энергией Q, которая теряется в виде теплоты. Однако из первого начала термодинамики нельзя определить часть расходуемой внутренней энергии, которая может быть преобразована в работу. Теоретические оценки затрат осуществляются на основе второго начала термодинамики. Этот закон накладывает строгие ограничения на эффективность преобразования энергии в работу и, кроме того, позволяет ввести критерии возможности самопроизвольного протекания того или иного процесса. Процесс называется самопроизвольным, если он осуществляется без каких-либо воздействий, когда система предоставлена самой себе. Существуют процессы, при которых внутренняя энергия системы не меняется (ДЕ = 0). К таким процессам относится, например, ионизация уксусной кислоты в воде. Целый ряд самопроизвольных процессов протекает с увеличением внутренней энергии (ДЕ > 0). Сюда относятся, в частности, типичные реакции образования бионеорганических соединений альбумина (белок плазмы крови) с ионами металлов, например Сu2+. Изменение внутренней энергии АЕ для закрытых систем не может служить критерием самопроизвольного протекания процессов. Следовательно, первого начала термодинамики, из которого получен этот критерий, недостаточно для решения вопроса о самопроизвольности, равно как и об эффективности процессов. Решение этих вопросов достигается с помощью второго начала термодинамики. Для формулировки второго начала термодинамики необходимо ввести понятия обратимого и необратимого в термодинамическом смысле процессов.Если система находится в равновесии, это состояние поддерживается как угодно долго при неизменности внешних условий. При изменении внешних условий состояние системы может меняться, т. е. в системе может протекать процесс. Процесс называется термодинамически обратимым, если при переходе из начального состояния 1 в конечное состояние 2 все промежуточные состояния оказываются равновесными. Процесс называется термодинамически необратимым, если хоть одно из промежуточных состояний не–равновесно. Обратимый процесс можно осуществить лишь при достаточно медленном изменении параметров системы – температуры, давления, концентрации веществ и др. Скорость изменения параметров должна быть такой, чтобы возникающие в ходе процесса отклонения от равновесия были пренебрежимо малы. Следует отметить, что с обратимостью связана важная проблема медицины – консервация тканей при низких температурах. Обратимые процессы являются предельным случаем реальных процессов, происходящих в природе и осуществляемых в промышленности или в лабораториях.
В качестве критерия самопроизвольности процессов в открытых и закрытых системах вводится новая функция состояния – энергия Гиббса. Эта функция получила название в честь великого американского физика Д. У. Гиббса (1839—1903), который вывел эту функцию, а затем использовал в термодинамических работах.Энергия Гиббса определяется через энтальпию Н и энтропию S с помощью соотношений:
G = H – S, ДG = ДH – ДS.
На основе энергии Гиббса второе начало термодинамики можно сформулировать следующим образом: в изобарно-изотермических условиях (р, Т = const) в системе самопроизвольно могут осуществляться только такие процессы, в результате которых энергия Гиббса системы уменьшается (ДG <0).В состоянии равновесия энергия Гиббса системы не ме–няется (G = const, AG = 0).
ДG < 0, р, Т = const. Из изложенного вытекает, что энергия Гиббса играет большую роль в изучении биоэнергетических процессов. С помощью этой функции состояния можно прогнозировать направление самопроизвольных процессов в биологических системах и рассчитывать мак-симально достижимый КПД.
Энергия Гиббса G так же, как и энтальпия Н, является функцией состояния системы. Поэтому изменение энергии Гиббса ДG может использоваться для характеристики химических превращений аналогично изменению энтальпии ДН. Уравнения реакции, для которых указывается соответствующее этим реакциям изменение энергии Гиббса, также называются термохимическими. Химические реакции, при протекании которых происходит уменьшение энергии Гиббса системы (ДG < 0) и совершается работа, называются экзергоническими. Реакции, в результате которых энергия Гиббса возрастает (ДG > 0) и над системой совершается работа, называются эндергоническими. Выведенная на основе второго начала термодинамики энергия Гиббса является функцией состояния. Следовательно, так же, как и для энтальпии, может быть сформулирован закон Гесса для энергии Гиббса в следующей форме: изменение энергии Гиббса при образовании заданных продуктов из данных реагентов при постоянных давлении и температуре не зависит от числа и вида реакций, в результате которых образуются эти продукты.Важный пример применения закона Гесса – расчет энергии Гиббса реакции окисления глюкозы дикислородом. Изменение энергии Гиббса в этой реакции при р = 101 кПа и Т = 298°К, определенное вне организма, равно ДG° = –2880 кДж/моль. Соответствующее термохимическое уравнение записывается в виде:
С6Н12О6 + 6О2 = 6СО2 + 6Н2О, ДGp-я° = –2880 кДж/моль.
Скорость химических реакций. Зависимость скорости реакции от концентрации. Закон действующих масс. Константа скорости реакции
Под скоростью химической реакции понимается изменение концентрации одного из реагирующих веществ в единицу времени. Концентрация вещества определяется числом молей в литре. При этом безразлично, о каком из участвующих в реакции веществ идет речь: псе они связаны между собой уравнениями реакции. По изменению концентрации одного из веществ можно судить о соответствующих изменениях концентраций всех остальных. При рассмотрении вопроса о скорости реакции необходимо различать гомогенные и гетерогенные реакции.Гомогенные реакции протекают в однородной среде, например в смеси газов или в водном растворе. Например:
2S02 + 02 -> 2SOsгаз газ газ
Гетерогенные реакции идут на поверхности соприкосновения твердого вещества и газа, твердого вещества и жидкости и т. д. Например:
Fe + 2HCl-> FeCl2 + H2t
В связи с этим скорость гомогенной реакции и скорость гетерогенной реакции определяются различно.Скорость гомогенной реакции математически можно отразить так:
V=+,- дельта с/дельта t
v — скорость реакции в гомогенной системе; с,, с2 — начальная и конечная концентрации одного из веществ, измеряемая числом молей в литре (моль/л);г,, t2 — моменты времени (секунды, минуты).Если речь идет о концентрации исходного вещества (концентрация которого с течением времени уменьшается), то берется знак «-». Если скорость оценить увеличением концентрации одного из продуктов реакции, то берется знак «+».Скорость реакции выражается в моль/л-с.Учитывая это, скорость гомогенной реакции можно определить так: Скорость гомогенной реакции определяется количеством вещества, вступающего в реакцию или образующегося при реакции за единицу времени в единице объема. Если реакция протекает между веществами в гетерогенной системе, то реагирующие вещества соприкасаются между собой не по всему объему, а только на поверхности. В связи с этим определение скорости гетерогенной реакции следующее. Скорость гетерогенной реакции определяется числом молей вещества, вступившего в реакцию или образующегося при реакции за единицу времени на единице поверхности. В математической формуле это можно отразить так:
v гетерог= дельта v / S дельта t
v — скорость гетерогенной реакции;
Av — количество молей вещества, вступившего или образующегося в реакции (моль);
S — площадь поверхности соприкосновения (м );
t — промежуток времени (секунда, минута).
Скорость реакции выражается в моль/м –с
Факторы, влияющие на скорость химической реакцииРассмотрим важнейшие факторы, которые влияют на скорость химической реакции. Скорость химической реакции зависит от следующих факторовПрирода реагирующих веществ. Под природой вещества понимают физическую природу, обусловленную строением его молекулы и взаимным влиянием атомов внутри ее, которое зависит от их электронного строения. Например, при равных условиях реакция более активного металла (цинка) с раствором соляной кислоты идет с бурным выделением водорода, менее активного (олово) — довольно медленно:
Zn + 2НС1 4 ZnCL, + Н2Т
Sn + 2HC1 =nCl2 + H2t
Концентрация реагирующих веществ. При увеличении концентрации реагирующих веществ число столкновений между их молекулами увеличивается, поэтому увеличивается и скорость реакции. Например, горение угля в чистом кислороде происходит быстрее, чем в воздухе, где содержание кислорода составляет 20-21%:с + о2 -> со2Площадь поверхности соприкосновения реагирующих веществ. Для твердых веществ скорость прямо пропорциональна поверхности реагирующих веществ, которую можно увеличить при измельчении и перемешивании.
Например, быстрее реагирует с соляной кислотой порошок алюминия, чем кусочек алюминия:
2А1 + 6НС1 =2А1С13 + ЗН2Т
Температура. При повышении температуры скорость большинства реакций увеличивается. Зависимость скорости реакции от температуры определяется правилом Вант-Гоффа: при повышении температуры на 10° скорость реакции увеличивается в 2-4 раза. Например, водород и кислород при комнатной температуре практически не взаимодействуют, а при 600°С реагируют мгновенно (взрыв):2Н2 + 02 Hi 2H20 Увеличение температуры приводит к увеличению числа активных молекул, обладающих достаточной энергией для взаимодействия.Катализатор. Катализаторами называют вещества, которые увеличивают скорости химических реакций, но сами при этом не расходуются. Явление такого ускорения реакций называется катализом. В зависимости от того, в одинаковых или различных агрегатных состояниях находится катализатор и реагирующие вещества, различают гомогенный и гетерогенный катализ. При гетерогенном катализе, когда катализатор представляет собой твердое вещество, а реакция проходит в растворе или газе, реагирующие вещества адсорбируются на поверхности катализатора, где и происходит собственно химическая реакция:
СН2=СН2 + Н2 =СН3-СН3этилен этан
N2 + 3H2=NH3 аммиак
При гомогенном катализе, когда катализатор и реагирующие вещества находятся в одной фазе, катализатор ускоряет реакцию путем образования промежуточных веществ при взаимодействии с одним из исходных компонентов, например:
2SO, + О., =2SO, (NO, - катализатор)
2 2 3 2
Все вещества в газовой фазе.
СН3СООН + С2Н2ОН -> СН3СООС2Н5 + Н20
уксусная этанол 2 4 этиловый эфир. кислота уксусной кислоты
Все реагенты в жидкой фазе.Особую роль играют биологические катализаторы — ферменты. При их участии протекают сложные химические процессы в растительных и животных организмах. Однако, кроме катализаторов, которые ускоряют реакции, есть вещества, которые замедляют химические реакции, то есть снижают скорость реакции. Их называют ингибиторами. Например соединения мышьяка, ртути, свинца, цианистые соединения и др.Влияние концентрации реагирующих веществ может быть объяснено из представлений, согласно которым химическое взаимодействие является результатом столкновения частиц. Увеличение числа частиц в заданном объеме приводит к более частым их столкновениям, т.е. к увеличению скорости реакции. Количественно зависимость между скоростью реакции и молярными концентрациями реагирующих веществ описывается основным законом химической кинетики — законом действующих масс. Скорость химической реакции при постоянной температуре прямо пропорциональна произведению концентраций реагирующих веществ.Для мономолекулярной реакции скорость реакции определяется концентрацией молекул вещества А:v = k*[A]где k — коэффициент пропорциональности, который называется константой скорости реакции; [А] — молярная концентрация вещества А.В случае бимолекулярной реакции, ее скорость определяется концентрацией молекул не только вещества А, но и вещества В:v = k*[A]*[B]В случае тримолекулярной реакции, скорость реакции выражается уравнением: v = k*[A]2*[B]В общем случае, если в реакцию вступают одновременно т молекул вещества А и n молекул вещества В, т. е.тА + пВ = С, уравнение скорости реакции имеет вид:v = k*[A]m*[B]nЭто уравнение есть математическое выражение закона действующих масс в общем виде. Чтобы понять физический смысл константы скорости реакции, надо принять в написанных выше уравнениях, что [А] = 1 моль/л и [В] = 1 моль/л (либо приравнять единице их произведение), и тогда v = k. Отсюда ясно, что константа скорости k численно равна скорости реакции, когда концентрации реагирующих веществ (или их произведение в уравнениях скорости) равны единице. Общее выражение для скорости химической реакции получено для данной, фиксированной температуры. В общем же случае, поскольку скорость реакции зависит от температуры, закон действующих масс записывается как v(T) = k(T) *[A]m*[B]n де v и k являются функциями температуры Константа скорости химической реакции, ее зависимость от температуры Многочисленные опыты показывают, что при повышении температуры скорость большинства химических реакций существенно увеличивается, причем для реакций в гомогенных системах при нагревании на каждые десять градусов скорость реакции возрастает в 2—4 раза (правило Вант-Гоффа).Это правило связано с понятием температурного коэффициента скорости реакции г и определяется соотношением г = kТ+10 / kТ Значение температурного коэффициента г дает возможность рассчитать изменение скорости реакции при увеличении температуры на некоторое число градусов от Т1 до Т2 по формуле v(Т1)/v(Т2) = г(Т2-Т1)/10Очевидно, что при повышении температуры в арифметической прогрессии скорость реакции возрастает в геометрической.
15 Скорость химических реакций. Порядок и молекулярность реакций
Под скоростью химической реакции понимается изменение концентрации одного из реагирующих веществ в единицу времени. Концентрация вещества определяется числом молей в литре.При этом безразлично, о каком из участвующих в реакции веществ идет речь: псе они связаны между собой уравнениями реакции. По изменению концентрации одного из веществ можно судить о соответствующих изменениях концентраций всех остальных.При рассмотрении вопроса о скорости реакции необходимо различать гомогенные и гетерогенные реакции.Гомогенные реакции протекают в однородной среде, например в смеси газов или в водном растворе. Например:
2S02 + 02 -> 2SOsгаз газ газ
Гетерогенные реакции идут на поверхности соприкосновения твердого вещества и газа, твердого вещества и жидкости и т. д. Например:
Fe + 2HCl-> FeCl2 + H2t
твердое раствор
В связи с этим скорость гомогенной реакции и скорость гетерогенной реакции определяются различно.Скорость гомогенной реакции математически можно отразить так:
V=+,- дельта с/дельта t
v — скорость реакции в гомогенной системе;с,, с2 — начальная и конечная концентрации одного из веществ, измеряемая числом молей в литре (моль/л);г,, t2 — моменты времени (секунды, минуты).Если речь идет о концентрации исходного вещества (концентрация которого с течением времени уменьшается), то берется знак «-». Если скорость оценить увеличением концентрации одного из продуктов реакции, то берется знак «+».Скорость реакции выражается в моль/л-с.Учитывая это, скорость гомогенной реакции можно определить так: Скорость гомогенной реакции определяется количеством вещества, вступающего в реакцию или образующегося при реакции за единицу времени в единице объема. Если реакция протекает между веществами в гетерогенной системе, то реагирующие вещества соприкасаются между собой не по всему объему, а только на поверхности. В связи с этим определение скорости гетерогенной реакции следующее. Скорость гетерогенной реакции определяется числом молей вещества, вступившего в реакцию или образующегося при реакции за единицу времени на единице поверхности. В математической формуле это можно отразить так:
v гетерог= дельта v / S дельта t
v — скорость гетерогенной реакции;
Av — количество молей вещества, вступившего или образующегося в реакции (моль);
S — площадь поверхности соприкосновения (м );
t — промежуток времени (секунда, минута).
Скорость реакции выражается в моль/м –с
Молекулярность и порядок реакцииПри изучении реакций выделяют молекулярность и порядок реакции.
Молекулярность реакции - это число молекул исходных веществ, принимающих участие в одном (единичном) химическом превращении. При этом число молекул образующихся продуктов не имеет значения. В соответствии с приведенным определением различают реакции:
1) мономолекулярные, в которых только один вид молекул участвует в превращении, причем стехиометрический коэффициент в уравнении равен единице, например,запись А > С означает, что молекула вещества А превращается в молекулу вещества С;
2) бимолекулярные, в которых участвуют два различных вида молекул или две молекулы одного вида (стехиометрический коэффициент во втором случае равен двум), например, А + В>С или 2А > С;
3) тримолекулярные, в которых участвуют три молекулы одного или разного видов, например,
А + В + D > С или 2 А + В > С, или 3А>С.
Реакции более высокой молекулярности маловероятны. Связано это с причиной, о которой говорилось ранее. Выше было сказано, что порядок химической реакции выражаетсяс уммой:
где аi - показатели степени концентрации исходных веществ в уравнении действующих масс.
Они приравнивались стехиометрическим коэффициентам компонентов химической реакции. Исходя из этого можно сделать заключение, что молекулярность и порядок реакции это одинаковые величины. Однако, это не всегда так. Порядок реакции или равен молекулярности или, в большинстве случаев, меньше её. Расхождение между порядком реакции и её молеклярностью может быть вызвано разными причинами.
1. Молекулярность реакции величина теоретическая, а порядок реакции - экспериментальная. Между теоретическими и экспериментальными величинами почти всегда есть различия.
2. Если, например, в реакции bB + dD = P,скорость которой W = КСBbCD d
один из компонентов, например, компонент B, находится в избытке, то в ходе данной реакции его концентрация будет изменяться незначительно и в уравнении скорости реакции можно принять СB = const. Но в таком случае скорость реакции практически зависит от концентрации только компонента D, то есть W = К1CD d тогда порядок реакции равен d, а молекулярность реакции (b + d).
3. Если данная реакция является гетерогенной, то в зависимости от условий протекания порядок такой реакции может быть различным.
4. Порядок каталитической реакции также может отличаться от молекулярности, причина - сложный механизм таких реакций.
5. Для сложной реакции, протекающей в несколько стадий, порядок реакции и её молекулярность не совпадают. В данном случае порядок реакции определяет какая-либо промежуточная (лимитирующая) стадия. Как правило порядок этой стадии отличается от молекулярности сложной реакции.
16. Зависимость скорости реакции от температуры. Правило Вант-Гоффа. Уравнение Аррениуса
ВАНТ-ГОФФА ПРАВИЛО. Почти все химические реакции при повышении температуры идут быстрее. Зависимость скорости реакции от температуры описывается уравнением Аррениуса:
k = Ae–Ea/RT,
где k – константа скорости реакции, А – не зависящая от температуры константа (ее называют предэкспоненциальным множителем), Еа – энергия активации, R – газовая постоянная, Т – абсолютная температура. В школьных учебниках зависимость скорости реакции от температуры определяют в соответствии с так называемым «правилом Вант-Гоффа», которое в 19 в. сформулировал голландский химик Якоб Вант-Гофф. Это чисто эмпирическое правило, т.е. правило, основанное не на теории, а выведенное из опытных данных. В соответствии с этим правилом, повышение температуры на 10° приводит к увеличению скорости в 2–4 раза. Математически эту зависимость можно выразить уравнением v2v1 = g (T2 – T1)/10, где v1 и v2 – скорости реакции при температурах Т1 и Т2; величина g называется температурным коэффициентом реакции. Например, если g = 2, то при Т2 – Т1 = 50о v2/v1 = 25 = 32, т.е. реакция ускорилась в 32 раза, причем это ускорение никак не зависит от абсолютных величин Т1 и Т2, а только от их разности.Однако из уравнения Аррениуса следует, что температурный коэффициент реакции зависит как от энергии активации, так и от абсолютной температуры. Для данной реакции с определенным значением Еа ускорение при повышении температуры на 10° будет тем больше, чем ниже температура. Это почти очевидно и без расчетов: повышение температуры от 0 до 10° С должно сказаться на скорости реакции значительно сильнее, чем такое же повышение температуры, например, от 500 до 510° С.
С другой стороны, для данного температурного интервала ускорение реакции будет тем сильнее, чем больше ее энергия активации. Так, если энергия активации реакции мала, то такая реакция идет очень быстро, и при повышении температуры на 10° С ее скорость почти не изменяется. Для таких реакций температурный коэффициент намного меньше 2. Для реакций же с большой энергией активации, которые при невысоких температурах идут медленно, ускорение при повышении температуры на 10° С может значительно превысить 4-кратное.Например, реакция диоксида углерода со щелочным раствором с образованием гидрокарбонат-иона (СО2 + ОН® НСО3–) имеет энергию активации 38,2 кДж/моль, поэтому при повышении температуры, например, от 50 до 60° С эта реакция ускорится всего в 1,5 раза. В то же время реакция распада этилбромида на этилен и бромоводород (С2Н5Вr ® С2Н4 + НВr) с энергией активации 218 кДж/моль ускорится при повышении температуры от 100 до 110oС в 6,3 раза (правда, в этом интервале температур реакция идет очень медленно). Кинетика реакции атомов водорода с этаном H + C2H6 ® H2 + C2H5 была изучены в широком температурном интервале – от 300 до 1100 К (27–827° С). Для этой реакции Eа = 40,6 кДж/моль. Следовательно, повышение температуры на 10° вызовет увеличение скорости реакции в 1,7 раза в интервале 300–310 K и только в 1,04 раза в интервале 1090–1100 K. Так что при высоких температурах скорость этой реакции практически не зависит от температуры. А для реакции присоединения атома водорода к двойной связи H + C2H4 ® C2H5 энергия активации мала (Eа = 3,4 кДж/моль, так что ее скорость слабо зависит от температуры в широком температурном интервале. И только при температурах намного ниже 0° С начинает сказываться наличие активационного барьера.
Подобных
примеров можно
привести множество.
Очевидно, что
правило Вант-Гоффа
противоречит
не только уравнению
Аррениуса, но
и многим экспериментальным
данным. Откуда
же оно взялось
и почему нередко
выполняется?Если
в приведенном
выше математическом
выражении для
правила Вант-Гоффа
подставить
вместо скоростей
v1 и v2 для данной
реакции их
зависимости
от температуры,
в соответствии
с уравнением
Аррениуса, то
после сокращения
предэкспоненциальных
множителей
получим следующее
выражение: g =
vT +10/vT = е–Еа/R(Т+10)/е–Еа/RТ
= е(Еа/R)[1/Т – 1/(T+10)].
Логарифмироване
этого уравнения
дает: lng = (Eа/R)[1/T – 1/(T +
10)], откуда Еа =
Rlng T(T + 10)/10 = 0,83lngT(T + 10). Энергия
активации не
является функцией
температуры,
эта зависимость
нужна лишь для
удобства последующего
анализа. Последнее
уравнение –
это уравнение
параболы, в
котором физический
смысл имеют
только положительные
значения.
Соответствующая
диаграмма
ограничена
двумя ветвями
параболы: при
g = 2 получаем Еа
= 0,58Т(Т + 10), при g = 4 получаем
Еа = 1,16Т(Т + 10). К тем
же формулам
приходим и при
использовании
десятичных
логарифмов.
Соответствующие
графики двух
парабол, для
значений g 2 и
4, приведены на
рисунке. Их
физический
смысл заключается
в том, что области
выполнения
правила Вант-Гоффа
соответствует
только область
между параболами.
Таким образом,
существуют
только определенные
соотношения
между энергией
активации
реакции и
температурой
ее проведения,
при которых
правило Вант-Гоффа
выполняется.
Ниже нижней
ветви температурный
коэффициент
g < 2, тогда как
выше верхней
ветви g > 4. Правило
Вант-Гоффа: при
повышении Т
на
![]() скорость хим.
реакции увеличивается
в 2-4 раза. Математически
это правило
можно записать:
скорость хим.
реакции увеличивается
в 2-4 раза. Математически
это правило
можно записать:
![]() ,
,
![]() ,
,
![]()
![]() -
температурный
коэффициент
хим. реакции.
Правило Вант-Гоффа
является приближённым
и его обычно
используют
для приблизительно
оценки скорости
при изменении
температуры.
Более точным
является уравнение
Аррениуса, по
которому:
-
температурный
коэффициент
хим. реакции.
Правило Вант-Гоффа
является приближённым
и его обычно
используют
для приблизительно
оценки скорости
при изменении
температуры.
Более точным
является уравнение
Аррениуса, по
которому:![]() .
Они могут быть
вычислены по
значению констант
скорости при
2-х различных
Т. При
.
Они могут быть
вычислены по
значению констант
скорости при
2-х различных
Т. При
![]() :
:
![]() (1). При
(1). При
![]() :
:
![]() (2). Вычитая из
(1) (2) получаем
(2). Вычитая из
(1) (2) получаем
![]() .
Отсюда можно
выразить А.
Зная А, по уравнению
(1) или (2) вычисляют
В. Уравнение
Аррениуса может
быть получено
т/д-им выводом
из уравнения
изобары (изохоры)
хим. реакции.
Опуская индексы,
характеризующие
условия протекания
реакции, это
уравнение
записывается:
.
Отсюда можно
выразить А.
Зная А, по уравнению
(1) или (2) вычисляют
В. Уравнение
Аррениуса может
быть получено
т/д-им выводом
из уравнения
изобары (изохоры)
хим. реакции.
Опуская индексы,
характеризующие
условия протекания
реакции, это
уравнение
записывается:
![]() ,
,
![]() ,
где
,
где
![]() и
и
![]() -
константы
скорости прямой
и обратной
реакции. Учитывая
эти уравнения
можно записать:
-
константы
скорости прямой
и обратной
реакции. Учитывая
эти уравнения
можно записать:
![]() .
Представим
тепловой эффект
реакции Q как
разность 2-х
энергетических
величин:
.
Представим
тепловой эффект
реакции Q как
разность 2-х
энергетических
величин:
![]() .
Тогда последнее
уравнение можно
записать в
виде:
.
Тогда последнее
уравнение можно
записать в
виде:
![]() .
С точностью
до некоторой
постоянной
величины можно
записать:
.
С точностью
до некоторой
постоянной
величины можно
записать:
![]() ,
,
![]() .
Опыт показывает
что
.
Опыт показывает
что
![]() .
Отбрасывая
индексы, последнее
уравнение
записывается:
.
Отбрасывая
индексы, последнее
уравнение
записывается:
![]() (1), где К – константа
скорости хим.
реакции. Энергетическая
величина Е в
этом уравнение
называется
энергией активации.
Полученное
уравнение
описывает
зависимость
К хим. реакции
от температуры.
Разделив переменные
и проинтегрировав,
получим:
(1), где К – константа
скорости хим.
реакции. Энергетическая
величина Е в
этом уравнение
называется
энергией активации.
Полученное
уравнение
описывает
зависимость
К хим. реакции
от температуры.
Разделив переменные
и проинтегрировав,
получим:
![]() ,
,
![]() (2).
(2).
Уравнение (2) по форме походит на уравнение Аррениуса, интегрируя (2) получим:
 ,
,
![]() (3).
(3).
Уравнение
используют
либо для вычисления
энергии активации
по известным
константам
скорости при
двух температурах,
либо для вычисления
константы
скорости реакции
при неизменной
температуре,
если известна
энергия активации.
Для большинства
хим. реакций
энергия активации
определяется
в пределах
![]() .
Физический
смысл энергии
активации
раскрывается
в теории химической
кинетики, её
можно определить
как некоторый
избыток энергии
по сравнению
со средним
значением для
денных условий,
которыми должны
обладать молекулы
чтобы вступить
в хим. реакцию.
Уравнение (2)
чаще представляют
в виде:
.
Физический
смысл энергии
активации
раскрывается
в теории химической
кинетики, её
можно определить
как некоторый
избыток энергии
по сравнению
со средним
значением для
денных условий,
которыми должны
обладать молекулы
чтобы вступить
в хим. реакцию.
Уравнение (2)
чаще представляют
в виде:
![]() .
При этом
.
При этом
![]() называют
предэкспоненциальным
множителем.
Связь энергии
активации с
тепловым эффектом
можно проиллюстрировать
с помощью
представлению
о энергетическом
барьере, который
разделяет
начальное и
конечное состояние
системы. I и II –
уровни энергии
вещ-в исходных
и продуктов
реакции.
называют
предэкспоненциальным
множителем.
Связь энергии
активации с
тепловым эффектом
можно проиллюстрировать
с помощью
представлению
о энергетическом
барьере, который
разделяет
начальное и
конечное состояние
системы. I и II –
уровни энергии
вещ-в исходных
и продуктов
реакции.
![]() -
энергия активации
прямой реакции.
-
энергия активации
прямой реакции.
![]() -
энергия активации
обратной реакции.
Избыток энергии
реагирующих
молекул, названный
энергией активации,
необходим для
преодоления
отталкивания
электронных
облаков взаимодействующих
молекул при
их столкновении,
и для разрыва
старых связей
молекул. Уравнение
Аррениуса
справедливо
в области невысоких
температур;
при достаточно
высоких температурах
константа
скорости перестаёт
зависеть от
температуры.
-
энергия активации
обратной реакции.
Избыток энергии
реагирующих
молекул, названный
энергией активации,
необходим для
преодоления
отталкивания
электронных
облаков взаимодействующих
молекул при
их столкновении,
и для разрыва
старых связей
молекул. Уравнение
Аррениуса
справедливо
в области невысоких
температур;
при достаточно
высоких температурах
константа
скорости перестаёт
зависеть от
температуры.
17. Энергия активации. Активированные комплексы. Уравнение Аррениуса
Энергия активации, разность между значениями средней энергии частиц (молекул, радикалов, ионов и др.), вступающих в элементарный акт химической реакции, и средней энергии всех частиц, находящихся в реагирующей системе. Для различных химических реакций Э. а. изменяется в широких пределах — от нескольких до ~ 10 дж./ моль. Для одной и той же химической реакции значение Э. а. зависит от вида функций распределения молекул по энергиям их поступательного движения и внутренним степеням свободы (электронным, колебательным, вращательным). Как статистическую величину Э. а. следует отличать от пороговой энергии, или энергетического барьера, — минимальной энергии, которой должна обладать одна пара сталкивающихся частиц для протекания данной элементарной реакции.В рамках представлений теории абсолютных скоростей реакций Э. а. — разность между значениями средней энергии активированных комплексов и средней энергии исходных молекул.Представления об Э. а. возникли в 70—80-х гг. 19 в. в результате работ Я. Вант-Гоффа и С. Аррениуса, посвященных изучению влияния температуры на скорость химической реакции. Константа скорости реакции k связана с Э. а. (Е) уравнением Аррениуса: k = koe-E/RT где R — газовая постоянная, Т — абсолютная температура в К, ko — постоянная, называемая предэкспоненциальным множителем константы скорости. Это уравнение, основанное на молекулярно-кинетической теории, позже было получено в статистической физике с учетом ряда упрощающих предположений, одно из которых — независимость Э. а. от температуры. Для практики и для теоретических расчетов в сравнительно узких температурных интервалах это предположение справедливо.
Э. а. можно найти по экспериментальным данным несколькими способами. Согласно одному из них, исследуют кинетику реакции при нескольких температурах (о методах см. в ст. Скорость химической реакции) и строят график в координатах In k — 1/T; тангенс угла наклона прямой на этом графике, в соответствии с уравнением Аррениуса, равен Е. Для одностадийных обратимых реакций (см. Обратимые и необратимые реакции) Э. а. реакции в одном из направлений (прямом или обратном) можно вычислить, если известна Э. а. реакции в другом и температурная зависимость константы равновесия (из термодинамических данных). Для более точных расчетов следует учитывать зависимость Э. а. от температуры. Э. а. сложных реакций представляет собой комбинацию Э. а. элементарных стадий. Иногда, помимо истинной Э. а., определяемой по уравнению Аррениуса, используют понятие "кажущейся" Э. а. Например, если константы скоростей гетерогенно-каталитических реакций определяют по изменению объемных концентраций исходных веществ и продуктов, то кажущаяся Э. а. отличается от истинной на величину тепловых эффектов, сопровождающих процессы адсорбции и десорбции реагирующих веществ на поверхности катализатора. В неравновесных системах, например плазмохимических (см. Плазмохимия), определение Э. а. является очень сложной задачей. В некоторых случаях, однако, возможно формальное применение уравнения Аррениуса. Э. а. — важнейшее понятие кинетики химической; ее значения включают в специальные справочники и используют в химической технологии для расчета скоростей реакций в различных условиях.
В теории активированного комплекса для любой элементарной реакции предполагается, что начальная конфигурация атомов переходит в конечную в результате непрерывного изменения межъядерных расстояний.Например в ходе элементарной реакции А + ВС -> АВ + С сближаются атомы А и В. Расстояние А-В (R1) уменьшается, а расстояние В-С (R2) увеличивается.Установлено, что подобные реакции осуществляются с наименьшей затратой энергии, если атомы располагаются на одной линии, соединяющей их центры. Тогда ход реакции можно описать, использую всего два межъядерных расстояния R1 и R2. В процессе непрерывного изменения межъядерных расстояний всегда образуется промежуточная конфигурация А...В...С, в которой связь В-С уже ослаблена, но еще не полностью разорвана, а связь А-В уже начала образовываться. Такая конфигурация является критической для данной реакции. Продукты реакции могут появиться только при условии образования этой конфигурации, которая называется переходным состоянием или активированным комплексом.
18 Скорость химических реакций. Катализ. Гомогенный и гетерогенный катализ
Под скоростью химической реакции понимается изменение концентрации одного из реагирующих веществ в единицу времени. Концентрация вещества определяется числом молей в литре.При этом безразлично, о каком из участвующих в реакции веществ идет речь: псе они связаны между собой уравнениями реакции. По изменению концентрации одного из веществ можно судить о соответствующих изменениях концентраций всех остальных.При рассмотрении вопроса о скорости реакции необходимо различать гомогенные и гетерогенные реакции.Гомогенные реакции протекают в однородной среде, например в смеси газов или в водном растворе. Например:
2S02 + 02 -> 2SOsгаз газ газ
Гетерогенные реакции идут на поверхности соприкосновения твердого вещества и газа, твердого вещества и жидкости и т. д. Например:
Fe + 2HCl-> FeCl2 + H2t
твердое раствор
В связи с этим скорость гомогенной реакции и скорость гетерогенной реакции определяются различно.Скорость гомогенной реакции математически можно отразить так:
V=+,- дельта с/дельта t
v — скорость реакции в гомогенной системе;с,, с2 — начальная и конечная концентрации одного из веществ, измеряемая числом молей в литре (моль/л);г,, t2 — моменты времени (секунды, минуты).Если речь идет о концентрации исходного вещества (концентрация которого с течением времени уменьшается), то берется знак «-». Если скорость оценить увеличением концентрации одного из продуктов реакции, то берется знак «+».Скорость реакции выражается в моль/л.
Катализом
называется
явление увеличения
скорости или
возбуждения
хим. р-ии происходящей
под действием
некоторых
веществ. Вещества,
в присутствии
которых наблюдается
указанное
явление называются
катализаторами.
Катализаторы,
участвуя в
р-ии, по её завершении
вновь регенерируются,
оставаясь
неизменными.
Различают
гомогенный,
когда катализатор
и реагирующие
вещества находятся
в одной фазе,
гетерогенный,
когда катализатор
и реагенты в
разных фазах,
кроме этого
выделяют
ферментативный
катализ, когда
катализатором
являются ферменты.
Микрогетерогенный
катализ, когда
катализатор
присутствует
в каллойдном
состоянии,
размер частиц
см. особенности:
1) катализатор
изменяет скорость
лишь таких
р-ии, которые
т/д возможны
при данных
условиях
![]() .
Прежде чем
подбирать
катализатор
для той или
иной р-ии нужно
убедиться в
её т/д возможности.
.
Прежде чем
подбирать
катализатор
для той или
иной р-ии нужно
убедиться в
её т/д возможности.
2) Катализатор непосредственно участвует в р-ях, образовывая промежуточные соединения, изменяя тем самым число и вид элементарных стадий процесса. Схематично можно представить так. Пусть для р-ии А+В=С+D катализатор является вещество К, тогда в присутствии катализатора схема может быть такой А+К→АК (а), АК+В=C+D+K (б). Р-ии (а) и (б) идут во много раз быстрее, чем р-ии непосредственного взаимодействия без катализатора. Промежуточное соединение в катализе это не обычное устойчивое хим. соединение, которое может быть выделено в чистом виде или может существовать в виде отдельной фазы. В гомогенном катализе это очень не стойкие соединения с малым временем жизни. В гетерогенном катализе это поверхностное соединение, не существующее в виде отдельной фазы, свойства которого резко отличаются от свойств аналогичного соединения образовывающего объёмную фазу.
3) Катализатор не изменяет величины теплового эффекта р-ии, в противном случае имело бы место не соблюдения з. сохранения и превращения энергии.
4) Катализатор не изменяет величины константы равновесия ∆H,∆S, ∆U, ∆F. Это означает, что равновесие, выход в присутствии катализатора остаётся тем же самым. Катализатор изменяет кинетические характеристики р-ии (EАКТ и предэкспоненты PZ0). действие катализатора может быть объяснено энергией активации процесса. Е3 и Е4 – энергии активации при образовании промежуточных соединений (а) и при его распаде (б), вследствие того, что энергия активации Е1 заменяет меньшую энергию Е3 и Е4 2-ух последних стадий скорость р-ии возрастает, даже если Е3+ Е4.>Е1 CH3CHO→CH4+CO при t=518єC ЕАКТ=45.5 ккал/моль в присутствии паров J2 энергия активации снижается до ЕАКТ=32,5 ккал/моль. Скорость возрастает в 10000 раз. Катализатор ускоряет и прямую и обратную р-ии в равной мере, при этом константа равновесия не изменяется. А время достижения системой равновесного состояния уменьшается.
5) Катализатор действует избирательно. Различные катализаторы могут или одну р-ию или группу р-ий или же р-ии различного класса в соответствии с этим катализаторы могут обладать индивидуальной специфичностью, групповой специфики или являться универсальными. Особое значение катализаторов имеет и используется при протекании в параллельных р-иях.
6) В гетерогенном катализе большое влияние на процесс имеет адсорбционная способность твердых катализатора, состояние его поверхности, способов и методов обработки поверхности, присутствие на поверхности атомов других элементов и т.д.
Например: введённые в катализатор некоторые добавки, которые сами не обладают каталитической активностью, могут сильно повысить активность катализатора, такие добавки называются промоторами. Наоборот присутствие некоторых др. веществ на поверхности катализатора может сильно снизить его каталитическую активность, такие вещества называются каталитическими ядрами.
Гомогенный катализ
Среди многочисленных каталитических реакций особое место занимает катализ в цепных реакциях.
“Цепными реакциями, как известно, называются такие химические и физические процессы, в которых образование в веществе или в смеси веществ некоторых активных частиц (активных центров) приводит к тому, что каждая из активных частиц вызывает целый ряд (цепь) последовательных превращений вещества” (Эмануэль, 1957).
Такой механизм развития процесса возможен благодаря тому, что активная частица взаимодействует с веществом, образуя не только продукты реакции, но и новую активную частицу (одну, две или более), способную к новой реакции превращения вещества, и т. д. Возникающая при этом цепь превращений вещества продолжается до тех пор, пока активная частица не исчезает из системы (происходит “гибель” активной частицы и обрыв цепи). Наиболее трудная стадия при этом - зарождение активных частиц (например, свободных радикалов), после же зарождения цепь превращений осуществляется легко.
Цепные реакции широко распространены в природе. Полимеризация, хлорирование, окисление и многие другие химические процессы идут по цепному, а точнее - по радикально-цепному (с участием радикалов) механизму. Механизм окисления органических соединений (на ранних стадрях) в настоящее время установлен достаточно тщательно. Если обозначить окисляющееся вещество R-H (где Н - атом водорода, имеющий наименьшую прочность связи с остальной молекулой R), то этот механизм можно записать в следующем виде: Катализаторы, например соединения металлов переменной валентности, могут оказывать влияние на любую из рассмотренных стадий процесса. Остановимся теперь на роли катализаторов в процессах вырожденного разветвления цепей. Взаимодействие гидроперекиси с металлом может приводить как к ускорению так и к торможению реакции окисления органических веществ соединениями металлов переменной валентности в зависимости от характера продуктов, образующихся при распаде гидроперекиси. Соединения металлов образуют с гидроперекисями комплекс, который распадается в “клетке” растворителя среды, если обра-зующиеся при распаде комплекса радикалы успеют выйти из клетки, то они инициируют процесс (положительный катализ). Если же эти радикалы не успеют выйти и рекомбинируют в клетке в молекулярные неактивные продукты, то это приведет к замедлению радикально-цепного процесса (отрицательный катализ), поскольку в этом случае гидроперекись - потенциальный поставщик новых радикалов- расходуется вхолостую. До сих пор мы рассматривали лишь неглубокие стадии процессов окисления; на более глубоких стадиях например в случае окисления углеводородов, образуются кислоты, спирты, кетоны, альдегиды, которые также могут реагировать с катализатором и служить дополнительным источником свободных радикалов в реакции, т. е. в этом случае будет налицо дополнительное вырожденное разветвление цепей.
Гетерогенный катализ
К сожалению, до сих пор, несмотря на достаточно большое число теорий и гипотез в области катализа, многие основополагающие открытия были сделаны случайно или в результате простого эмпирического подхода. Как известно, случайно был найден ртутный катализатор сульфирования ароматических углеводородов М. А. Ильинским, который нечаянно разбил ртутный термометр: ртуть попала в реактор, и реакция пошла. Аналогичным образом были обнаружены теперь всем хорошо известные, а в свое время открывшие новую эру в процессе полимеризации катализаторы стереоспецифической полимеризации Циглера. Естественно, что такой путь развития учения о катализе не соответствует современному уровню науки, и именно этим объясняется повышенный интерес к изучению элементарных стадий процессов в гетерогенно-каталитических реакциях. Эти исследования - прелюдия для создания строго научных основ подбора высокоэффективных катализаторов.
Во многих случаях роль гетерогенных катализаторов в процессе окисления сводится к адсорбции органического соединения и кислорода с образованием на поверхности катализатора адсорбированного комплекса этих веществ. Такой комплекс разрыхляет связи компонентов и делает их более реакционноспособными. В некоторых случаях катализатор адсорбирует лишь один компонент, который диссоциирует на радикалы. Например, пропилен на закиси меди диссоциирует с образованием аллильного радикала, легко вступающего затем в реакцию с кислородом.
Выяснилось, что каталитическая активность металлов переменной валентности в значительной мере зависит от заполнения d-орбиталей в катионах окислов металлов.
По каталитической- активности в реакции разложения многих гидроперекисей соединения металлов располагаются следующим ря-
Мы рассмотрели один из возмжных путей инициирования процесса - взаимодействие гидроперекиси с катализатором. Однако в случае окисления реакция гетерогенного инциирования цепей может протекать как путем распада на радикалы гидроперекиси, так и путем взаимодействия углеводорода с кислородом, активированным поверхностью катализатора. Инициирование цепей может быть обусловлено участием заряженной формы органического соединения RH+, образующегося при взаимодействии RH с катализатором. Так обстоит дело с катализом в реакциях инициирования (зарождения и разветвления) цепей. Роль гетерогенных катализаторов в реакциях продолжения цепи особенно четко подчеркивается изменением скорости и направления изомеризации перекисных радикалов.
19 Химическое равновесие. Факторы, влияющие на смещение равновесия. Принцип Ле Шателье. Константа химического равновесия, ее связь с термодинамическими функциями
Химические реакции заключаются во взаимодействии реагентов с образованием продуктов. Не следует, однако полагать, что направление химической реакции только одно. В действительности, химические реакции протекают и в прямом, и в обратном направлениях. Реагенты n Продукты. Такие реакции называются обратимыми. Обратимые (и необратимые) химические реакции бывают как гомогенными, так и гетерогенными. Гомогенными называют реакции, протекающие в одной фазе – газовой или жидкой. Они характеризуются отсутствием поверхности раздела между реагентами и продуктами, взаимодействие которых осуществляется во всем объеме реакционной смеси. У гомогенных обратимых реакций все участники находятся в одном агрегатном состоянии и составляют одну фазу, например:
Реакция между газообразными веществами
СН4(г) + СО2(г) n 2СО(г) + 2Н2(г)
Реакция этирификации, протекающая в водной среде.Равновесие, имеющее место у таких реакций, называется гомогенным.Обратимые гомогенные и гетерогенные реакции в отличие от необратимых идут не конца, т.е. не до полного исчезновения реагентов: они «прекращаются» прежде, чем будут полностью израсходованы их исходные вещества (если они были взяты в стехиометрических соотношениях), поэтому в реакционной смеси у таких реакций всегда присутствуют и исходные вещества и продукты их взаимодействия. максимальный выход продуктов у них менее 100% и соответствует равновесному составу. Такие реакции протекают до установления в них определенного концентрационного предела, общего для их прямого и обратного направлений, называемого состоянием химического равновесия. Именно с наступлением последнего и связывают прекращение протекания реакции в целом. Равновесие в системе наступает в результате стремления ее к минимальному значению энергии и максимальному хаотическому распределению в пространстве, характеризующемуся энтропией. Достигнутое и неизменное при состоянии химического равновесия соотношение достаточно больших, отчетливо определяемых аналитически концентраций всех веществ называют положением химического равновесия.
Одна из важнейших количественных характеристик химического равновесия – константа равновесия, позволяющая судить о полноте протекания реакции при тех или иных условиях. В общем случае применительно к любым химическим системам, как идеальным, так и реальным, константа равновесия Кравн обратимой химической реакции есть величина постоянная при данных температуре, давлении и в данном растворителе.Для гомогенных химических равновесий, устанавливающихся в идеальных жидких и газообразных (газовых смесях) растворах, константу равновесия можно выразить через равновесные молярные концентрации и равновесные молярные доли, а для равновесий в газовых смесях – через равновесные парциальные давления: (для реакции nАА(Г) + nВВ(Г) n nD D(Г) + n F F(Г)
Кравн = [D] nD [F] n F = КС - моль/м3
[А] nА [В] n В
Кравн =Хравн DnD Хравн F n F = КХ – безразмерная величина
ХравнА nА Хравн В n В
Кравн = Рравн DnD Рравн F n F = КР – Па (атм)
РравнА nА Рравн В n В
Коэффициенты активности и фугитивности компонентов таких систем равны единице, поэтому активности становятся равными концентрациям, а фугитивности – давлениям.
Активностью (фугитивностью) называют величину, при подстановке которой вместо концентрации (парциального давления) в выражения, выведенные для идеальных систем, можно применить их к реальным системам.Константы КС, КХ, КР иногда называют концентрационными или эмпирическими константами равновесия, поскольку для их расчета используются экспериментально определяемые значения равновесной молярной концентрации (в квадратных скобках), парциальные давления компонентов рравн i, равновесные молярные доли Хравн i.
Кр = Кс (RТ)rnг ; Кр = Кх робщrnг ; Кс = Кх Vм-rnг где Vм мольный объем смеси (раствора);
rnг = (nD + n F ) – (nА + n В)
– изменение числа молей газообразных веществ, рассчитываемое по разности между суммами стехиометрических коэффициентов продуктов и реагентов. (стр 331 -333 А.А.Гуров)
Связь между константами равновесия и термодинамическими характеристиками системы устанавливается уравнениями изотермы, изобары и изохоры химической реакции. Эти уравнения впервые были выведены Вант-Гоффом (1886г), поэтому в литературе их часто называют его именем, а именно: изотерма, изобара и изохора Вант-Гоффа. Каждое из них имеет несколько разновидностей и используется либо в дифференциальной, либо в интегральной формах. Уравнение изотермы в общем виде связывает энергию Гиббса, константу равновесия и начальные (или текущие), т.е. неравновесные концентрации (активности в случае реальных растворов) или неравновесные парциальные давления (фугитивности в случае реальных газовых смесей) всех компонентов химической реакции.
(rr G)р,Т = - RТ ln Кр0 + R Т ln рDn D р F n F
рАn А р В n В
Которое и называют уравнением изотермы химической реакции в общем виде.
По уравнению изотермы вычисляют энергию Гиббса реакции, а также определяют направление и глубину ее самопроизвольного протекания при данных условиях, если известны стандартная константа равновесия для данной температуры и исходный состав произвольно приготовленной реакционной смеси, т.е. неравновесные относительные, например, парциальные давления рi всех компонентов в начальный момент времени. Очевидно, что чем больше абсолютное значение (rr G)р,Т, тем менее равновесна система.
-если в реакционной смеси присутствуют или только продукты реакции или только исходные вещества, то (rr G)р,Т ®±Ґ для любой реакции;
- (rr G)р,Тс 0 реакция протекает в обратном направлении справа налево до установления равновесия;
-(rr G)р,Тб 0 реакция протекает самопроизвольно в прямом направлении;
-(rr G)р,Т= 0 отвечает состоянию химического равновесия;
-Если величина, стоящая под знаком логарифма во втором слагаемом равна единице (это возможно, например при начальных относительных парциальных давлениях всех компонентов, равных единице, т.е. компоненты находятся в стандартных условиях), то само слагаемое равно нулю, а уравнение изотермы трансформируется:
(rr G)р,Т = - RТ ln Кр0 или rr GТ0 = - RТ ln Кр0
и его называют стандартным уравнением изотермы химической реакции или уравнением стандартного сродства. С его помощью можно вычислить Кр0 без экспериментального изучения равновесия.
Уравнение изобары химической реакции определяет зависимость константы равновесия Кр0 от температуры и в дифференциальной форме выводится на основании стандартного уравнения изотермы
rr GТ0 = - R ln Кр0
Т
и дифференциального уравнения энергии Гиббса – Гельмгольца для стандартных условий
rr GТ0 = rr НТ0 + Т d (rr GТ0)
d Т
После преобразований получают уравнение изобары:
d ln Кр0 = rr Н0Т
d Т RТ2
Анализируя его можно установить, что, чем больше абсолютная величина теплового эффекта и чем ниже температура, тем больше значение производной, следовательно, сильнее сказывается изменение температуры на константе равновесия и в большей степени смещается положение равновесия. Уравнение полезно при вычислении графическим путем тепловых эффектов реакции. Если температурная зависимость константы равновесия выражена в координатах ln Кр0 - 1/ Т.
Газофазная реакция осуществляется в закрытой системе, при постоянном объеме (V = соnst).
Уравнение изохоры химической реакции в дифференциальной форме:
d ln К с0 = rr U0Т
d Т RТ2
В интегральной форме:
ln К 0с2 = rr U0Т (Т2-Т1)
К 0с1 RТ1 Т2
Где rr U0Т - изменение стандартной внутренней энергии реакции; К с0 - безразмерная стандартная константа равновесия, выраженная через относительное равновесные молярные концентрации компонентов.
К внешним факторам, влияющим на состояние равновесия любой системы (гомогенной или гетерогенной), относятся температура, давление, внешние поля (электрическое, магнитное, гравитационное и др.). Если значение последнего фактора существенно не отличается от нормального им пренебрегают. Обычно рассматривают воздействие только двух важных факторов: температуры, давления.
Влияние изменения внешних условий на состояние равновесия устанавливается термодинамическим положением, впервые сформулированным Брауном, которое называется правилом смещения положения подвижного равновесия, или принципом Ле Шателье: если на систему, находящуюся в состоянии истинного химического равновесия, оказывать внешнее воздействие путем изменения какого-либо из условий (Сi, Т, рi. Робщ), определяющих положение равновесия, то в системе происходит изменение равновесного состава и смещение положения равновесия в направлении того процесса, протекание которого ослабляет эффект (влияние) этого воздействия
20 Растворы. Физическая и химическая теории растворов
Растворами называются гомогенные системы, содержащие не менее двух веществ. Могут существовать растворы твердых, жидких и газообразных веществ в жидких растворителях, а также однородные смеси (растворы) твердых, жидких и газообразных веществ. Как правило, вещество, взятое в избытке и в том же агрегатном состоянии, что и сам раствор, принято считать растворителем, а компонент, взятый в недостатке - растворенным веществом.В зависимости от агрегатного состояния растворителя различают газообразные, жидкие и твердые растворы.Газообразными растворами являются воздух и другие смеси газов.К жидким растворам относят гомогенные смеси газов, жидкостей и твердых тел с жидкостями.Твердыми растворами являются многие сплавы, например, металлов друг с другом, стёкла. Наибольшее значение имеют жидкие смеси, в которых растворителем является жидкость. Наиболее распространенным растворителем из неорганических веществ, конечно же, является вода. Из органических веществ в качестве растворителей используют метанол, этанол, диэтиловый эфир, ацетон, бензол, четыреххлористый углерод и др.В процессе растворения частицы (ионы или молекулы) растворяемого вещества под действием хаотически движущихся частиц растворителя переходят в раствор, образуя в результате беспорядочного движения частиц качественно новую однородную систему. Способность к образованию растворов выражена у разных веществ в различной степени. Одни вещества способны смешиваться друг с другом в любых количествах (вода и спирт), другие - в ограниченных (хлорид натрия и вода).
Сущность процесса образования раствора можно показать на примере растворения твердого вещества в жидкости. С точки зрения молекулярно-кинетической теории растворение протекает следующим образом: при внесении в растворитель какого-либо твердого вещества, например, поваренной соли, частицы ионов Na+ и Cl-, находящиеся на поверхности, в результате колебательного движения, увеличивающегося при соударении с частицами растворителя, могут отрываться и переходить в растворитель. Этот процесс распространяется на следующие слои частиц, которые обнажаются в кристалле после удаления поверхностного слоя. Так постепенно частицы, образующие кристалл (ионы или молекулы), переходят в раствор. На рис дана наглядная схема разрушения ионной кристаллической решетки NaСl при растворении в воде, состоящей из полярных молекул. Частицы, перешедшие в раствор, вследствие диффузии распределяются по всему объему растворителя. С другой стороны, по мере увеличения концентрации частицы (ионы, молекулы), находящиеся в непрерывном движении, при столкновении с твердой поверхностью еще не растворившегося вещества могут задерживаться на ней, т.е. растворение всегда сопровождается обратным явлением - кристаллизацией. Может наступить такой момент, когда одновременно выделяется из раствора столько же частиц (ионов, молекул), сколько их переходит в раствор - наступает равновесие. По соотношению преобладания числа частиц, переходящих в раствор или удаляющихся из раствора, различают растворы насыщенные, ненасыщенные и пересыщенные. По относительным количествам растворенного вещества и растворителя растворы подразделяют на разбавленные и концентрированные.Раствор, в котором данное вещество при данной температуре больше не растворяется, т.е. раствор, находящийся в равновесии с растворяемым веществом, называют насыщенным, а раствор, в котором еще можно растворить добавочное количество данного вещества, - ненасыщенным.Насыщенный раствор содержит максимально возможное (для данных условий) количество растворенного вещества. Следовательно, насыщенным раствором является такой раствор, который находится в равновесии с избытком растворенного вещества. Концентрация насыщенного раствора (растворимость) для данного вещества при строго определенных условиях (температура, растворитель) - величина постоянная.Раствор, содержащий растворенного вещества больше, чем его должно быть в данных условиях в насыщенном растворе, называется пересыщенным. Пересыщенные растворы представляют собой неустойчивые, неравновесные системы, в которых наблюдается самопроизвольный переход в равновесное состояние. При этом выделяется избыток растворенного вещества, и раствор становится насыщенным.
Насыщенный и ненасыщенный растворы нельзя путать с разбавленным и концентрированным. Разбавленные растворы - растворы с небольшим содержанием растворенного вещества; концентрированные растворы - растворы с большим содержанием растворенного вещества. Необходимо подчеркнуть, что понятие разбавленный и концентрированный растворы являются относительными, выражающими только соотношение количеств растворенного вещества и растворителя в растворе.
Сравнивая растворимость различных веществ, мы видим, что насыщенные растворы малорастворимых веществ являются разбавленными, а хорошо растворимых веществ - хотя и ненасыщенные, но довольно концентрированными.В зависимости от того, электронейтральными или заряженными частицами являются компоненты раствора, их подразделяют на молекулярные (растворы неэлектролитов) и ионные (растворы электролитов). Одна из характерных особенностей растворов электролитов заключается в том, что они проводят электрический ток. Химическая теория растворов развивалась Д.И.Менделеевым, который рассматривал растворы как смеси непрочных химических соединений молекул растворенного вещества с молекулами растворителя. Эти представления основывались на экспериментальной зависимости плотности водно-спиртовых растворов от состава и на зависимости первой производной от плотности спиртового раствора по числу моль спирта в зависимости от состава раствора.
Развитием теории Д.И.Менделеева является полиэдрическая теория образования растворов, согласно которой в жидкости из однородных и разнородных молекул создаются элементарные пространственные группы-полиэдры. Однако химическая теория не может объяснить механизм образования идеальных растворов, отклонения в свойствах реальных растворов от свойств идеальных растворов и др.
Для объяснения свойств идеальных растворов с изменением их состава била предложена физическая теория (В.Ф.Алексеев - 1870--1880 гг.) и была проложена в работах Вант-Гоффа, Аррениуса и др. Согласно этой теории, процесс растворения одного вещества в другом является результатом простого распределения молекул по объему тепловым движением, при этом между взаимодействующими молекулами проявляются только слабые MМB. В результате простого перемешивания молекул веществ процесс растворения при создании идеальных растворов не сопровождается тепловым эффектом. Идеальными свойствами обладают также разбавленные растворы. Идеальными свойствами в любых пределах изменения концентрации растворов обладают смеси веществ с близкими по свойствам молекулами: н-гептан-н-гексан, оптические изомеры.Общая теория растворов в настоящее время не создана, хотя проводятся широкие научные исследования методами квантовой химии, статистической термодинамики, кристаллохимии, различными физико-химическими методами анализа Д.И.Менделеев указывал, что образование растворов может рассматриваться с двух сторон: с физической и химической, и в растворах виднее, чем где-либо, насколько эти стороны естествознания сближены между собою. В учении о растворах широко используются представления о полиэдрической их структуре и развиваются аналитические методы, связывающие структуру, состав и свойства растворов.
21. Фазовые равновесия в гетерогенных системах, фазовые превращения и правило фаз. Диаграммы состояния
Фазовое равновесие, сосуществование термодинамически равновесных фаз гетерогенной системы. Является одним из основных случаев термодинамического равновесия и включает в себя условия равенства температуры всех частей системы (термическое равновесие), равенства давления во всем объеме системы (механическое равновесие) и равенство химических потенциалов каждого компонента во всех фазах системы, что обеспечивает равновесное распределение компонентов между фазами. Число фаз f, находящихся одновременно в равновесии, связано с числом компонентов k, числом n независимых параметров, определяющих состояние системы (обычно, когда учитывается только влияние температуры и давления, n = 2), и числом термодинамических степеней свободы v уравнением: v = k + 2 - f (см. Фаз правило).В общем виде условие фазовое равновесие, согласно принципу равновесия Гиббса, сводится к максимуму энтропии S системы при постоянстве внутренней энергии U, общего объема V и числа молей каждого компонента ni-. Этот принцип можно выразить также как условие минимума любого из термодинамических потенциалов: внутренней энергии U, энтальпии H, энергии Гиббса G, энергии Гельмгольца А при условии постоянства соответствующих параметров состояния, включая число молей каждого компонента.Фазовые равновесия могут быть стабильными и метастабильными. Те и другие являются локально устойчивыми, то есть устойчивыми по отношению к малым возмущениям параметров состояния - температуры, давления, состава (концентраций компонентов). Метастабильные фазовые равновесия отличаются тем, что они неустойчивы к некоторым конечным изменениям этих параметров, ведущим, в частности, к переходу к другим фазам. Например, пересыщенный раствор или переохлажденный расплав неустойчивы по отношению к кристаллической фазе. Поскольку метастабильное состояние системы локально устойчиво, переход к стабильному состоянию требует преодоления некоторого активационного барьера и протекания процесса зародышеобразования (см. Зарождение новой фазы).
Следует отметить некоторые особенности метастабильных фаз: при одной и той же температуре давление пара выше над метастабильной фазой, чем над стабильной; при одном и том же давлении температура плавления метастабильной фазы ниже, чем стабильной; растворимость метастабильной фазы при постоянных давлении и температуре выше, чем стабильной. Последнее справедливо как для жидких, так и для твердых растворов.Критерий достижения фазового равновесия. Наиболее общий критерий достижения фазового равновесия - сходимость значений CB-B системы при их измерении, если подходить к состоянию фазового равновесия сверху (со стороны более высоких температур) и снизу (со стороны низких температур). Достижение фазового равновесия или хотя бы приближение к нему - важнейший вопрос при изучении диаграмм состояния, в том числе диаграмм растворимости, диаграмм плавкости, диаграмм давления пара, а также в физико-химическом анализе. При исследовании растворимости для достижения фазового равновесия применяют длительную (от нескольких часов до нескольких месяцев) выдержку образца с перемешиванием в термостате. В случае образования в системе твердых растворов рекомендуется подход к равновесию сверху, от более высоких температур, сочетающий быстрое охлаждение с целью получения мелких кристаллов и интенсивное перемешивание. При исследовании систем методом термического анализа обычно используют образцы, полученные сплавлением компонентов с последующим медленным охлаждением. В случае образования в системе твердых растворов и инконгруэнтно плавящихся фаз, а также фаз, разлагающихся в твердом состоянии, требуется проведение предварительного отжига образца при фиксированной температуре - от нескольких часов до нескольких месяцев. Для ускорения отжига сплавленных образцов рекомендуется предварительное быстрое охлаждение расплава.
При изучении твердых тел. состоящих из тугоплавких или разлагающихся при высоких температурах компонентов, применяют такие методы подготовки образцов, как прессование таблеток смесей перед отжигом и промежуточное перетирание смесей при отжиге, отжиг смесей солей или гелей, осажденных из водных или других растворов и т. п.Типы фазовых равновесий. В однокомпонентной системе (при наличии полиморфных превращений) возможны 4 вида двухфазных равновесий: жидкость - пар, кристалл - пар, кристалл - жидкость и кристалл - кристалл; 4 вида трехфазных равновесий: кристалл - жидкость - пар, кристалл - кристалл - жидкость, кристалл - кристалл - пар и кристалл - кристалл - кристалл; при этом не учитывается возможность образования жидких кристаллов. В двойных системах (компоненты А и В) возможны те же виды двухфазных равновесий, но число возможных видов трехфазных равновесий достигает 26 вследствие того, что играет роль не только природа сосуществующих фаз (их агрегатное состояние), но и взаимное расположение фазовых полей на диаграмме состояния в координатах температура - состав (давление предполагается постоянным). Все эти фазовые равновесия делятся на два типа: эвтектическое фазовое равновесие, при которых из трех одновременно участвующих в равновесии фаз при понижении температуры одна испытывает превращение, а две другие при этом образуются, и перитектическое фазовое равновесие, когда две фазы взаимодействуют (превращаются), при этом образуется третья фаза.
В простейшем случае, если на основе компонентов А и В возможно образование жидкого раствора L и двух твердых растворов a и b, эвтектического и перитектического фазовых равновесий можно записать соответственно в виде реакций:
![]()
Поскольку в двойной системе состояние трехфазного равновесия является нонвариантным, эвтектические и перитектические реакции происходят при постоянной температуре, называемой соотв. эвтектической или перитектической, то есть на диаграмме состояния этим равновесиям отвечают горизонтали. В случае, если в определенной области температур и составов все три равновесно сосуществующие фазы являются твердыми (у одного из компонентов существуют полиморфные модификации с образованием твердого раствора g), возможны трехфазные равновесия, называют эвтектоидными и перитектоидными. Их можно представить соответствующими реакциями, аналогично эвтектическим и перитектическим фазовым равновесиям:
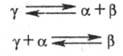
При наличии в некотором температурно-концентрационном интервале двух жидких фаз L1 и L2 и одной твердой (напр., а) возможны трехфазные равновесия, называют монотектическое и синтектическое:
Некоторые виды трехфазных равновесий, например те, при которых образуется жидкость в результате взаимодействием двух кристаллических фаз при понижении температуры, теоретически возможны, но реально, по-видимому, не наблюдаются. При переходе к тройным и более сложным системам число видов многофазных фазовых равновесий возрастает еще больше (см. Тройная точка).Распределение компонентов между фазами системы при фазовом равновесии описывается законом распределения, устанавливающим, что отношение термодинамических активностей примеси в двух фазах при фазовое равновесие является постоянной величиной. В первом приближении активности компонентов можно заменить их концентрациями. Одним из условий выполнимости закона распределения вещества между фазами является одинаковость молекулярного состояния растворенного вещества в обеих фазах, то есть отсутствие ассоциации молекул. Замена активностей на концентрации допустима, если коэффициент активности компонента в обеих фазах не зависят от концентрации, то есть для идеальных растворов (это условие обычно выполняется для очень разбавленных растворов, в случае микроконцентраций). Отношение активностей компонентов называют коэффициентом распределения или коэффициентом относительной летучести и т. п. Частные случаи закона распределения - правила и законы, выражающие равновесное распределение вещества в двухфазных системах. Например, для расчета равновесия жидкости и пара пользуются законами Рауля и Генри, первым - для вещества, находящегося в избытке, вторым - для вещества, являющегося примесью. Распределение растворенного вещества между двумя несмешивающимися жидкостями при постоянной температуре характеризуется тем, что отношение его концентраций в этих двух фазах сохраняется постоянным (закон Бертло - Нернста). Распределение примеси между жидкой и твердой кристаллической фазой описывается распределениями Хлопина (равновесия) и Дёрнера-Хоскинса.Законы распределения являются основой разнообразных гетерогенных методов очистки (разделения), хотя само фазовое равновесие в процессе проведения этих методов очистки достигается далеко не всегда, а иногда сама возможность очистки обусловлена отсутствием фазового равновесия (см. Кристаллизационные методы разделения смесей, Ректификация, Экстракция жидкостная).
Диаграммы состояния
Диаграммы состояния, или диаграммы фазового равновесия в удобной графической форме показывают фазовый состав сплава в зависимости от температуры и концентрации. Диаграммы состояния строят для условий равновесия (окончательное состояние). Равновесное состояние соответствует минимальному значению свободной энергии. Это состояние может быть достигнуто только при очень малых скоростях охлаждения или длительном нагреве. Однако истинное равновесие достигается редко, наиболее часто системы находятся в метастабильном состоянии (неустойчивом), и под воздействием внешних факторов могут переходить в другие более устойчивые состояния. Метастабильные состояния нередко сообщают сплавам высокие механические и другие свойства.
Правило фаз
Диаграммы фазового равновесия характеризуют окончательное состояние сплавов, то есть после того как все превращения в них произошли и полностью закончились. Это состояние зависит от внешних условий (Т0 С; Р, МПа) и характеризуется числом и концентрацией образовавшихся фаз. Закономерность изменения числа фаз в гетерогенной системе определяется правилом фаз.
Правило фаз устанавливает зависимость между числом степеней свободы, числом компонентов и числом фаз и выражается уравнением
С = К - Ф + 2,
где С - число степеней свободы системы (или вариантность);
К - число компонентов, образующих систему;
2 - число внешних факторов (Т и Р);
Ф - число фаз, находящихся в равновесии.
Под числом степеней свободы (вариантностью системы) понимают возможность изменения температуры, давления и концентрации без изменения числа фаз, находящихся в равновесии.
При нормальных условиях изменяется только один фактор -Т0С, Р =const, тогда:
С=К - Ф + 1.
Число степеней свободы не может быть меньше нуля, тогда К-Ф+1>0, а Ф<К+1, то есть число фаз в сплаве, не может быть больше чем число компонентов плюс единица. Таким образом, в двойной системе может быть не более трёх фаз.При С = 0 - существует в равновесии сразу три фазы - имеется нонвариантное равновесие (безвариантное). При таком равновесии сплав может существовать только при условии - постоянная температура и определённый состав всех фаз, находящихся в равновесии. То есть кристаллизация (или превращение) начинается и заканчивается при постоянной температуре. Если С =1 или 2, то кристаллизация или превращение протекает с течением времени в интервале температур.
Построение диаграмм состояния
Диаграмма состояния показывает изменение состояния сплавов в зависимости от температуры (P = const) и концентрации.
Если в системе имеется два компонента, то диаграмма будет иметь два измерения: первое - температурная шкала, второе - концентрация сплава (рисунок 2).
Рисунок 2.
Каждая точка на оси абсцисс соответствует определённому содержанию каждого компонента. Общее содержание компонентов в сплаве - 100%.
Крайние ординаты на диаграмме соответствуют чистым компонентам, а ординаты между ними - двойным сплавам.
Через точку С проходит сплав содержащий 35% компонента В и, соответственно, 65% компонента А.
Каждая точка на диаграмме состояния показывает состояние сплава данной концентрации при данной температуре. Каждая вертикаль соответствует изменению температуры определенного сплава. Изменение фазового состояния сплава отмечается на диаграмме точкой.
Линии, соединяющие точки аналогичных превращений, разграничивают на диаграмме области аналогичных фазовых состояний.
Вид диаграммы состояния зависит от того, как реагируют оба компонента друг с другом в твердом и жидком состоянии, то есть, растворимы ли они в жидком и твердом состоянии, образуют ли химические соединения и так далее.
Обычно диаграммы состояния строят, экспериментально используя термический анализ, то есть строят кривые охлаждения и по остановкам и перегибам на этих кривых, вызванным тепловым эффектом превращений, определяют температуры превращений. Эти температуры называют критическими точками.
Температуру металлов измеряют обычно при помощи термопары.
Имея достаточное количество сплавов, и определив в каждом сплаве температуры превращений, можно построить диаграмму состояния.
Рисунок 3. Построение кривых охлаждения
Диаграмма состояния показывает, какую структуру будет иметь медленно охлажденный сплав при любой температуре.
Растворы как многокомпонентные системы. Способы выражения состава растворов. Молярная доля, массовая доля. Молярная концентрация, молярная концентрация эквивалентов, моляльная концентрация
Растворы – это однородные (гомогенные) дисперсные системы, состоящие из двух или большего числа компонентов (относительные количества которых могут меняться в широких пределах) и продуктов их взаимодействия.
Растворы занимают промежуточное положение между механическими смесями (растворы характеризуются непостоянством своего состава и могут быть разделены на составные части) и химическими соединениями (растворы однородны, устойчивы, образование растворов сопровождается энергетическим эффектом).
В настоящее время установлено, что при растворении молекулы растворяемого вещества связываются с молекулами растворителя, при этом образуются сольваты (если растворитель вода, то образуются гидраты). На разрушение связей между молекулами энергия затрачивается, а при образовании гидрата (сольвата) энергия выделяется; разница между этими энергиями будет наблюдаться в виде теплового эффекта растворения, которая может быть как положительной, так и отрицательной.
Способы выражения концентрации растворов.
1) Массовая доля раствора щ (х). Выражается отношением массы растворенного вещества m(х) к массе раствора.
![]()
Является величиной безразмерной или выражается в процентах:
![]()
Например, 15%-ный раствор: массовая доля щ (х) = 0,15
2) Молярная концентрация раствора С(х). Выражается отношением количества растворенного вещества n(x) к объему раствора, выраженному в литрах.
![]()
Т.к. количество вещества n(x) выражается отношением массы вещества m(x) к его молярной массе M(x), то молярную концентрацию раствора удобно выразить как
![]()
Концентрация — величина, характеризующая количественный состав раствора.
Согласно правилам ИЮПАК, концентрацией растворённого вещества (не раствора) называют отношение количества растворённого вещества или его массы к объёму раствора (моль/л, г/л), то есть это соотношение неоднородных величин.
Те величины, которые являются отношением однотипных величин (отношение массы растворённого вещества к массе раствора, отношение объёма растворённого вещества к объёму раствора) правильно называть долями. Однако на практике для обоих видов выражения состава применяют термин концентрация и говорят о концентрации растворов.
Существует много способов выражения концентрации растворов. Массовая доля (также называют процентной концентрацией)
Массовая доля — отношение массы растворённого вещества к массе раствора. Массовая доля измеряется в долях единицы.
![]()
где:
m1 — масса растворённого вещества, г ;
m — общая масса раствора, г.
Массовое процентное содержание компонента, m%
m%=(mi/Уmi)*100
Молярность (молярная объёмная концентрация)
Молярная концентрация — количество растворённого вещества (число молей) в единице объёма раствора. Молярная концентрация в системе СИ измеряется в моль/мі, однако на практике её гораздо чаще выражают в моль/л или ммоль/л. Также распространено выражение в «молярности». Возможно другое обозначение молярной концентрации CM, которое принято обозначать М. Так, раствор с концентрацией 0,5 моль/л называют 0,5-молярным. Примечание: термин «моль» не склоняется по падежам. После цифры пишут «моль», подобно тому, как после цифры пишут «см», «кг» и т. д.
![]() где:
где:
н — количество растворённого вещества, моль;
V — общий объём раствора, л.
Эквивалентная концентрация сeqB растворенного вещества В – это отношение эквивалентного количества вещества neqB к объему раствора:
сeqB = neqB /V(p).
Единица эквивалентной концентрации – моль/л.
Раствор, в котором эквивалентная концентрация растворенного вещества равна сeqB (моль/л), характеризуется нормальноcтью, численно равной значению сeqB; обозначение нормальности раствора – н. (поcле числа). Например, раствор с эквивалентной концентрацией серной кислоты сeq(H2SO4) = 1 моль/л. может быть обозначен как 1 н. H2SO4 (однонормальный раствор серной кислоты в воде).
Эквивалентная концентрация растворенного вещества и нормальность раствора определяются эквивалентным количеством растворенного вещества и, следовательно, как и последнее, зависят от эквивалентного числа, постоянного только для конкретной реакции, причем сeqB = zBсB
Моляльная концентрация Cml (моль/кг) - это отношение количества растворенного вещества n к массе растворителя msol:
Cml = n
-----
msol
Моляльная концентрация не зависит от температуры раствора, т.к. масса раствора при различных температурах остается постоянной; а объем раствора, изменяется с изменением его температуры.
Обратите внимание, на то, что несмотря на созвучность в названиях: молярная и моляльная - это различные способы выражения концентрации растворенного вещества в растворе
Способы выражения состава раствора Важнейшей характеристикой раствора является его состав. Состав определяет раствор как в качественном отношении (из каких компонентов состоит раствор), так и в количе-ственном (в каких относительных количествах тот или иной компонент содержится в растворе).Существует много способов выражения количественного состава. Рассмотрим основные из них.
1. Молярная доля(хi) - отношение количества вещества (моль) компонента ni, содержащегося в данной системе, к общему числу молей в системе Eni.
Xi =ni/Еni. (11.1)
2. Объемная доля (фi) - отношение объема компонента (Vi), содержащегося в систе-ме, к общему объему V:
(Pi = Vi,/ V (11.2)
3. Массовая доля (a>i) - отношение массы компонента mi к массе смеси Emi.
coi = mi /Emi. (11.3)
4.Молярная концентрация (ci) - отношение количества вещества ni (моль), содержащегося в системе, к объему этой системы V:
Ci = ni/ V (11.4)
5.Молярностъ растворенного вещества (bi) - отношение количества растворенно-го компонента ni к массе вещества растворителя mp. Обычно рассматривают содержание в 1000 г растворителя.(bi) = ni /mp [моль/кг] (11.5)
6.Молярная концентрация эквивалента (cif) - отношение количества эквивалентоврастворенного вещества nif к объему системы V
ci = nif / V (11.6)
Свойства растворов неэлектролитов. Явление осмоса. Закон Вант-Гоффа. Давление насыщенного пара растворителя над раствором. Первый и второй законы Рауля
Первый закон Рауля связывает давление насыщенного пара над раствором с его составом; он формулируется следующим образом:
Парциальное
давление насыщенного
пара компонента
раствора прямо
пропорционально
его мольной
доле в растворе,
причём коэффициент
пропорциональности
равен давлению
насыщенного
пара над чистым
компонентом.
![]() Для бинарного
раствора, состоящего
из компонентов
А и В (компонент
А считаем
растворителем)
удобнее использовать
другую формулировку:
Для бинарного
раствора, состоящего
из компонентов
А и В (компонент
А считаем
растворителем)
удобнее использовать
другую формулировку:
Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.
![]()
Растворы, для которых выполняется закон Рауля, называются идеальными. Идеальными при любых концентрациях являются растворы, компоненты которых очень близки по физическим и химическим свойствам (оптические изомеры, гомологи и т. п.), и образование которых не сопровождается изменением объёма и выделением либо поглощением теплоты. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором. Отклонения от закона РауляРастворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области очень малых концентраций; при больших концентрациях наблюдаются отклонения от закона Рауля. Случай, когда истинные парциальные давления паров над смесью больше, чем вычисленные по закону Рауля, называют положительными отклонениями. Противоположный случай, когда парциальные давления паров компонентов оказываются меньше вычисленных — отрицательные отклоненияПричиной отклонений от закона Рауля является то обстоятельство, что однородные частицы взаимодействуют друг с другом иначе, чем разнородные (сильнее в случае положительных и слабее в случае отрицательных отклонений).Реальные растворы с положительными отклонениями от закона Рауля образуются из чистых компонентов с поглощением теплоты (ДНраств > 0); объём раствора оказывается больше, чем сумма исходных объёмов компонентов (ДV > 0). Растворы с отрицательными отклонениями от закона Рауля образуются с выделением теплоты (ДНраств < 0); объём раствора в этом случае будет меньше, чем сумма исходных объёмов компонентов (ДV < 0).
Второй закон Рауля
Тот факт, что давление паров над раствором отличается от давления паров над чистым растворителем, существенно влияет на процессы кристаллизации и кипения. Из первого закона Рауля выводятся два следствия, касающиеся понижения температуры замерзания и повышения температуры кипения растворов, которые в объединённом виде известны как второй закон Рауля. Понижение температуры кристаллизации растворов
Условием кристаллизации является равенство давления насыщенного пара растворителя над раствором давлению пара над твёрдым растворителем. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, это равенство всегда будет достигаться при температуре более низкой, чем температура замерзания растворителя. Так, океанская вода начинает замерзать при температуре около минус 2 °C.
Разность между температурой кристаллизации растворителя T°fr и температурой начала кристаллизации раствора Tfr есть понижение температуры кристаллизации.
Понижение температуры кристаллизации бесконечно разбавленных растворов не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора.
![]()
Поскольку по мере кристаллизации растворителя из раствора концентрация последнего возрастает, растворы не имеют определённой температуры замерзания и кристаллизуются в некотором интервале температур. Повышение температуры кипения растворов
Жидкость кипит при той температуре, при которой общее давление насыщенного пара становится равным внешнему давлению. Если растворённое вещество нелетуче (то есть давлением его насыщенных паров над раствором можно пренебречь), то общее давление насыщенного пара над раствором равно парциальному давлению паров растворителя. В этом случае давление насыщенных паров над раствором при любой температуре будет меньше, чем над чистым растворителем, и равенство его внешнему давлению будет достигаться при более высокой температуре. Таким образом, температура кипения раствора нелетучего вещества Tb всегда выше, чем температура кипения чистого растворителя при том же давлении T°b.
Повышение температуры кипения бесконечно разбавленных растворов нелетучих веществ не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора
![]() .
.
РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ, бинарные или многокомпонентные мол. системы, состав к-рых может изменяться непрерывным образом (по крайней мере, в нек-рых пределах). В отличие от растворов электролитов, в растворах неэлектролитов (мол. р-рах) заряженные частицы в сколько-нибудь заметных концентрациях отсутствуют. Растворы неэлектролитов могут быть твердыми, жидкими и газообразными. В данной статье рассматриваются жидкие р-ры; см. также Твердые растворы.Взаимная р-римость двух жидкостей при заданных т-ре Т и давлении р м. б. полной (неограниченной) или ограниченной. В последнем случае р-ры в нек-рой области составов расслаиваются, т. е. разделяются на две жидкие фазы, отличающиеся по концентрации. В многокомпонентных расслаивающихся р-рах число сосуществующих жидких фаз м. б. более двух. Если один (или более) из компонентов раствора неэлектролитов в чистом состоянии при заданных Т и р является газом или твердым телом, область существования раствора неэлектролитов простирается от чистой жидкости (смеси жидкостей), выступающей в роли р-рителя, до состава, отвечающего насыщ. р-ру.Растворы неэлектролитов служат средой, в к-рой протекают многие прир. и пром. процессы. Изучение и прогнозирование св-в этих систем тесно связаны с такими практич. проблемами, как подбор р-рителей для реализации технол. процессов, получение систем с заданными св-вами, разделение прир. и пром. смесей (включая газы и нефти), глубокая очистка в-в.
Физ. химия изучает широкий диапазон св-в р-ров. Наиб. разработана и имеет практически важные применения равновесная термодинамика р-ров; дальнейший материал посвящен в осн. этому разделу физ. химии р-ров. Кроме того, изучаются транспортные св-ва р-ров-диффузия, теплопроводность, вязкость (см. Физико-химическая гидродинамика), а также спектроскопич., электрич., акустич. и др. физ. св-ва. Методы исследования макроскопич. св-в растворов неэлектролитов и их структурных характеристик во многом аналогичны методам исследования индивидуальных жидкостей, но осн. внимание уделяется рассмотрению концентрац. зависимостей св-в. Важнейшая задача физ.-хим. исследований - установление связи между наблюдаемыми на опыте св-вами, структурой р-ров и характеристиками межмолекулярных взаимодействий. Эксперим. информацию о структуре р-ров и межмолекулярных взаимод. в них дают методы оптической и радиоспектроскопии, дифракционные, электрич. и др. Важную роль в изучении растворов неэлектролитов играет физико-химический анализ, основанный на построении и исследовании фазовых диаграмм, концентрац. зависимостей термодинамич. и др. физ. св-в (показателя преломления, вязкости, теплопроводности, акустич. характеристик и др.). При этом одна из главных задач состоит в том, чтобы на основании анализа диаграмм состав - свойство устанавливать факт образования хим. соединений между компонентами растворов неэлектролитов и находить их характеристики.Значит. влияние на физ. св-ва р-ров (в частности, на рассеяние света) оказывают флуктуации плотности, концентрации, ориентации молекул. Роль флуктуации концентрации особенно велика вблизи критич. точки р-римости (см. Критические явления).Концентрационные зависимости термодинамических функций. Особенностью термодинамич. описания растворов неэлектролитов по сравнению с чистыми компонентами является наличие дополнит. термодинамич. степеней свободы системы, связанных с возможностью изменения состава системы (см. Фаз правило). Число степеней свободы гомогенного n-компонентного р-ра равно n+1. В качестве переменных, определяющих его состояние, наиб. удобно выбрать давление р, т-ру Т и концентрации п — 1 компонентов. Состав растворов неэлектролитов чаще всего выражают через молярные доли компонентов xi, считая независимыми переменными молярные доли всех компонентов, кроме n-го x1,..., xn-1. Для задания концентрации используют и др. шкалы (молярности с, моляльности т).
При описании концентрац. зависимостей термодинамич. ф-ций важную роль играют парциальные молярные величины Mi для i-го компонента, определяемые соотношением:
![]()
где М-любая экстенсивная термодинамич. ф-ция (объем V, внутр. энергия U, энтальпия H, энтропия S, энергии Гельм-гольца и Гиббса F и G, теплоемкость Ср и т.д.), mi-число молей. Важнейшая парциальная молярная величина -химический потенциал mi (парциальная молярная энергия Гиббса); именно через хим. потенциалы формулируются условия хим. и фазового равновесий в системе.
Концентрац.
зависимость
термодинамич.
св-в растворов
неэлектролитов
нередко характеризуют
функциями
смешения Мт
- изменением
термодинамич.
ф-ции М при
образовании
р-ра из чистых
жидкостей.
Рассматривают
смешение при
изотермо-изобар-ных
(Т, р = const) или изотермо-изохорных
(Т, V = const) условиях,
причем наиб.
практич. интерес
представляет
случай Т, р —
const. Молярная ф-ция
смешения при
этих условиях
(![]() )
определена
соотношением:
)
определена
соотношением:
![]()
где
![]()
молярное значение ф-ции M для чистой жидкости i при заданных Т и р. В частности, молярная энергия Гиббса смешения
![]()
где
![]() хим. потенциал
чистой жидкости
i при заданных
Т и р. Для чистых
жидкостей
хим. потенциал
чистой жидкости
i при заданных
Т и р. Для чистых
жидкостей
![]()
О́смос (от греч. ὄумпт «толчок, давление») — процесс односторонней диффузии через полупроницаемую мембрану молекул растворителя в сторону большей концентрации
Явление осмоса наблюдается в тех средах, где подвижность растворителя больше подвижности растворённых веществ. Важным частным случаем осмоса является осмос через полупроницаемую мембрану. Полупроницаемыми называют мембраны, которые имеют достаточно высокую проницаемость не для всех, а лишь для некоторых веществ, в частности, для растворителя. (Подвижность растворённых веществ в мембране стремится к нулю). Если такая мембрана разделяет раствор и чистый растворитель, то концентрация растворителя в растворе оказывается менее высокой, поскольку там часть его молекул замещена на молекулы растворенного вещества (см. Рис. 1). Вследствие этого, переходы частиц растворителя из отдела, содержащего чистый растворитель, в раствор будут происходить чаще, чем в противоположном направлении. Соответственно, объём раствора будет увеличиваться (а концентрация уменьшаться), тогда как объём растворителя будет соответственно уменьшаться.
Например, к яичной скорлупе с внутренней стороны прилегает полупроницаемая мембрана: она пропускает молекулы воды и задерживает молекулы сахара. Если такой мембраной разделить растворы сахара с концентрацией 5 и 10 % соответственно, то через нее в обоих направлениях будут проходить только молекулы воды. В результате в более разбавленном растворе концентрация сахара повысится, а в более концентрированном, наоборот, понизится. Когда концентрация сахара в обоих растворах станет одинаковой, наступит равновесие. Растворы, достигшие равновесия, называются изотоническими.
Осмос, направленный внутрь ограниченного объёма жидкости, называется эндосмосом, наружу — экзосмосом. Перенос растворителя через мембрану обусловлен осмотическим давлением. Оно равно избыточному внешнему давлению, которое следует приложить со стороны раствора, чтобы прекратить процесс, то есть создать условия осмотического равновесия. Превышение избыточного давления над осмотическим может привести к обращению осмоса — обратной диффузии растворителя.В случаях, когда мембрана проницаема не только для растворителя, но и для некоторых растворённых веществ, перенос последних из раствора в растворитель позволяет осуществить диализ, применяемый как способ очистки полимеров и коллоидных систем от низкомолекулярных примесей, например электролитов.
Значение осмосаОсмос играет важную роль во многих биологических процессах. Мембрана, окружающая нормальную клетку крови, проницаема лишь для молекул воды, кислорода, некоторых из растворенных в крови питательных веществ и продуктов клеточной жизнедеятельности; для больших белковых молекул, находящихся в растворенном состоянии внутри клетки, она непроницаема. Поэтому белки, столь важные для биологических процессов, остаются внутри клетки.
Осмос участвует в переносе питательных веществ в стволах высоких деревьев, где капиллярный перенос не способен выполнить эту функцию.Осмос широко используют в лабораторной технике: при определении молярных характеристик полимеров, концентрировании растворов, исследовании разнообразных биологических структур. Осмотические явления иногда используются в промышленности, например при получении некоторых полимерных материалов, очистке высоко-минерализованной воды методом «обратного» осмоса жидкостей.Клетки растений используют осмос также для увеличения объёма вакуоли, чтобы она распирала стенки клетки (тургорное давление). Клетки растений делают это путём запасания сахарозы. Увеличивая или уменьшая концентрацию сахарозы в цитоплазме, клетки могут регулировать осмос. За счёт этого повышается упругость растения в целом. С изменениями тургорного давления связаны многие движения растений (например, движения усов гороха и других лазающих растений). Пресноводные простейшие также имеют вакуоль, но задача вакуолей простейших заключается лишь в откачивании лишней воды из цитоплазмы для поддержания постоянной концентрации растворённых в ней веществ.
Осмос также играет большую роль в экологии водоёмов. Если концентрация соли и других веществ в воде поднимется или упадёт, то обитатели этих вод погибнут из-за пагубного воздействия осмоса.
Вант-Гоффа закон
Вант-Гоффа закон осмотического давления, определяет давление молекул растворённого вещества на полупроницаемую перепонку, отделяющую раствор от чистого растворителя и непроницаемую для растворённого вещества
Давление насыщенного пара является важным свойством раствора.
Если к чистому растворителю с давлением насыщенного пара р0 добавить постороннее нелетучее вещество, то давление пара растворителя над раствором изменится. При растворении какого-либо нелетучего вещества в данном растворителе понижается концентрация молекул последнего в единице объема жидкости, и уменьшается число молекул, вылетающих в единицу времени из жидкой фазы в парообразную. При меньшей концентрации пара, т.е. при меньшем его давлении, установится равновесие.
Следовательно, давление насыщенного пара растворителя над раствором всегда меньше, чем над чистым растворителем. Понижение давления насыщенного пара растворителя над раствором тем больше, чем выше концентрация растворенного вещества. Для идеальных растворов давление насыщенного пара определяется законом Рауля, установленного им в 1886 г.: относительное понижение давления насыщенного пара растворителя над раствором равно молярной доле растворенного вещества, т.е. отношению количества данного вещества к общему количеству растворителя и растворенного вещества:
Др / р0 = р0 - р/р0 = n2 / n1 + n2 = х2, где
р0 - давление пара растворителя над чистым растворителем,
р - давление пара растворителя над раствором,
Др / р0 - относительное понижение давления пара растворителя,
n2 - количество растворенного вещества,
n1 - количество вещества растворителя,
х2 - молярная доля растворенного вещества.
Это уравнение можно представить в другом виде и придать закону иную формулировку:
![]()
давление пара над раствором равно произведению давления пара над чистым растворителем на молярную долю растворителя.
Для очень разбавленных растворов уравнение имеет вид:
![]()
m1 и m2 - массы растворителя и растворенного вещества соответственно,
М1 и М2 - молярные массы растворителя и растворенного вещества соответственно.
2) Понижение давления насыщенного пара влечет за собой понижение температуры замерзания раствора по сравнению с чистым растворителем.
Жидкость замерзает при той температуре, при которой давление насыщенного пара над ней такое же, как и над кристаллами этого вещества. Так как давление насыщенного пара растворителя над раствором всегда меньше, чем над чистым растворителем, то разбавленный раствор будет замерзать при более низкой температуре, чем растворитель. Температурой замерзания раствора считают ту температуру, при которой в процессе охлаждения начинают выделяться первые кристаллы чистого растворителя. Для таких растворов Рауль нашел, что понижение температуры замерзания раствора Дtз. = t0 - t (t0 - температура замерзания растворителя, t - температура замерзания раствора) пропорционально его моляльности (1 моль в 1000 г растворителя):
Дtз = К · m, где
Дt - понижение температуры замерзания,
m - моляльность раствора,
К - криоскопическая постоянная ("криос" - холод).
Физический смысл криоскопической постоянной К состоит в том, что она равна понижению tз. раствора, содержащего 1 моль растворенного вещества на 1 кг растворителя. Величина К зависит от природы растворенного вещества, если его молекулы не диссоциируют и не ассоциируют.
Величину К можно рассчитать по формуле
![]()
T0 -температура замерзания растворителя,
l пл - удельная теплота плавления растворителя.
Так как
![]() [моль/кг], то
[моль/кг], то
![]()
m1 - масса растворителя,
m2 - масса растворенного вещества.
3) Повышение температуры кипения.
Жидкость закипает при температуре, при которой давление насыщенного пара жидкости становится равным внешнему давлению. Так как давление насыщенного пара растворов нелетучих или малолетучих веществ меньше давления насыщенного пара растворителя, то эти растворы кипят при более высокой температуре, чем растворитель. Для разбавленных растворов таких веществ Рауль установил, что повышение температуры кипения раствора Дtк. = t - t0 пропорционально его моляльности:
Дtк. = Е · m, где
Е - эбуллиоскопическая постоянная ("эбуллиос" - кипеть),
m - моляльность раствора.
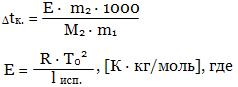
T0 -температура кипения растворителя,
lисп. - удельная теплота испарения растворителя. Величина Е численно равна повышению tк моляльного раствора при условии сохранения свойств раствора до этой концентрации. Метод определения молярной массы на основе измерения понижения температуры замерзания Дtз растворов - метод криоскопии, на основе измерения повышения температуры кипения Дtк. - эбуллиоскопический метод. Более распространен первый метод, так как Дtк растворов обычно мала и на нее влияют такие факторы как изменение внешнего давления, перегрев раствора и т.д. Одним из важных свойств растворов является осмос. Если отделить раствор от растворителя мембраной, проницаемой лишь для частиц растворителя, то будет наблюдаться самопроизвольный переход растворителя в раствор. Явление самопроизвольной диффузии растворителя через полупроницаемую перегородку в раствор называется осмосом. Сила, рассчитанная на единицу площади мембраны, заставляющая переходить растворитель через полупроницаемую перегородку в раствор, называется осмотическим давлением. По определению Вант-Гоффа осмотическое давление разбавленных растворов численно равно произведению молярной концентрации и температуры:
р = с · R · T,
где с - молярная концентрация.
Так как с = n / v, где n - количество вещества (моль), v - объем раствора (л), то
р = n / v · R · T,
Зависимость осмотического давления от понижения давления пара растворителя над раствором выражается уравнением:
![]()
где М0 - молярная масса растворителя. Уравнение Вант-Гоффа распространяется только на разбавленные растворы. При изучении свойств растворов электролитов (кислоты, щелочи, соли) было обнаружено, что этим растворам присущи все основные свойства растворов неэлектролитов, такие, как понижение давления насыщенного пара над раствором, понижение температуры замерзания, повышение температуры кипения и осмотическое давление. Однако эти величины для растворов электролитов оказались иными, чем следовало ожидать, исходя из их молярных концентраций.
Вант-Гофф, чтобы применить законы растворов неэлектролитов к электролитам, ввел поправочный множитель, названный изотоническим коэффициентом i. Этот коэффициент показывал, во сколько раз наблюдаемое осмотическое давление (рtз., рtк.) больше вычисленного, т.е.
р = i · с · R · T
Дtз = i · К · m
и т.д.
Для растворов неэлектролитов i = 1, для электролитов - i > 1.
В 1887 году С. Аррениус выдвинул гипотезу, согласно которой в растворах электролитов молекулы распадаются на ионы - катионы и анионы. Очень скоро эта гипотеза превратилась в теорию электролитической диссоциации (ЭДС). Основные положения ее таковы:
Вещества, распадающиеся на ионы в растворах или расплавах, и потому проводящие электрический ток, называются электролитами.
Электролиты при растворении в воде распадаются на ионы: положительные - катионы и отрицательные - анионы.
Диссоциация - процесс для большинства электролитов обратимый: наряду с распадом протекает процесс соединения ионов (ассоциация).
Водные растворы электролитов условно делятся на три группы: сильные, средние и слабые. Всвязи с чем введена величина степень диссоциации б = nдис. / nобщ., где nдис. - число молекул электролита, распавшихся на ионы, nобщ. - общее число молекул электролита в растворе.
Если б > 30% - это сильные электролиты, б = 5-30% имеют средние электролиты, б < 5% - слабые электролиты.
24.Растворы электролитов. Изотонический коэффициент. Теория электролитической диссоциации. Степень электролитической диссоциации. Понятие об активности
Электролитическая диссоциация, полный или частичный распад молекул растворенного вещества на катионы и анионы. Электролитической диссоциацией называют также распад на катионы и анионы ионных кристаллов при растворении или расплавлении. Электролитическая диссоциация, как правило, происходит в полярных растворителях.
При
электролитической
диссоциации
разрываются
обычно лишь
наиболее полярные
связи молекул,
например карбоновые
кислоты RCOOH диссоциируют
на
![]() и Н+, электролитической
диссоциации
могут подвергаться
молекулы некоторых
растворителей,
например воды.
и Н+, электролитической
диссоциации
могут подвергаться
молекулы некоторых
растворителей,
например воды.
Основными причинами электролитической диссоциации являются, с одной стороны, взаимодействие растворенного вещества с растворителем, которое приводит к сольватации ионов, а с другой стороны - значительное ослабление электростатических взаимодействий между сольватированными ионами в среде, обусловленное ее электростатическим полем (диэлектрической проницаемостью растворителя). При этом работа, необходимая для разрушения молекул (кристаллической решетки), обеспечивается за счет энергии сольватации. Электролитическая диссоциация лежит в основе деления растворов на два класса - растворы неэлектролитов и растворы электролитов. Наблюдаемое различие в коллигативных свойствах разбавленных растворов электролитов и неэлектролитов объясняется тем, что из-за электролитической диссоциации увеличивается общее число частиц в растворе. Это, в частности, приводит к увеличению осмотического давления раствора сравнительнос растворами неэлектролитов, понижению давления пара растворителя над раствором, увеличениюизменения температуры кипения и замерзания раствора относительно чистого растворителя. Электролитической диссоциацией объясняется также ионная электропроводность электролитов.
Мерой
электролитической
диссоциации
является степень
диссоциации
альфа- отношение
кол-ва диссоциированных
на ионы молекул
электролита
к их исходному
количеству
в растворе.
Согласно этому
определению
альфа- изменяется
от 0 (отсутствие
диссоциации)
до 1 (полная
диссоциация)
и зависит от
природы растворенного
вещества и
растворителя,
а также от
концентрации
раствора и
температуры.
Как правило,
с увеличением
диэлектрической
проницаемости
растворителя
![]() его
его
![]() увеличивается,
хотя заметная
диссоциация
наблюдается
в некоторых
растворителях
с низкой
увеличивается,
хотя заметная
диссоциация
наблюдается
в некоторых
растворителях
с низкой
![]() .
Способность
данного вещества
MX к электролитическая
диссоциация
в определенном
р-рителе по
схеме MX
.
Способность
данного вещества
MX к электролитическая
диссоциация
в определенном
р-рителе по
схеме MX
![]() M+
+ Х- характеризуется
константой
электролитической
диссоциации
KD, связанной,
согласно закону
действующих
масс, со степенью
диссоциации
альфа соотношением:
M+
+ Х- характеризуется
константой
электролитической
диссоциации
KD, связанной,
согласно закону
действующих
масс, со степенью
диссоциации
альфа соотношением:
![]()
где
х: - молярная
концентрация
электролита![]() -
средний ионный
коэффициент
активности;
-
средний ионный
коэффициент
активности;
![]() коэффициент
активности
недиссоциированной
части электролита.
Как и значение
константы KD
зависит от
свойств растворенного
вещества, в
частности от
прочности связи
между фрагментами
молекул электролита,
образующими
катион и анион,
от диэлектрических
свойств растворителя,
его способности
сольватировать
ионы, а также
от температуры
и давления; в
отличие от
альфа не зависит
от концентрации
раствора. Константа
KD может быть
определена
экспериментально,
например, по
зависимости
электропроводности
раствора от
концентрации
электролита
или путем прямого
измерения
содержания
свободных ионов
в растворе,
например,
спектрофотометрическим
методом.
коэффициент
активности
недиссоциированной
части электролита.
Как и значение
константы KD
зависит от
свойств растворенного
вещества, в
частности от
прочности связи
между фрагментами
молекул электролита,
образующими
катион и анион,
от диэлектрических
свойств растворителя,
его способности
сольватировать
ионы, а также
от температуры
и давления; в
отличие от
альфа не зависит
от концентрации
раствора. Константа
KD может быть
определена
экспериментально,
например, по
зависимости
электропроводности
раствора от
концентрации
электролита
или путем прямого
измерения
содержания
свободных ионов
в растворе,
например,
спектрофотометрическим
методом.
Соответственно
понятиям полной
и неполной
электролитической
диссоциации
электролиты
классифицируют
на сильные
![]() и слабые
и слабые
![]() (см.
Электролиты),
полностью
диссоциируют
в растворе
многие соли
неорганических
кислот, некоторые
кислоты и основания.
Неполная
электролитическая
диссоциация
наблюдается
для солей, катионы
которых склонны
к образованию
ковалентных
связей с анионами,
например соли
Ag, Cd, Zn. Некоторые
многоосновные
кислоты, например
H2SO4, полностью
диссоциируют
лишь в отношении
отщепления
одного иона
Н+, а дальнейшая
диссоциация
(см.
Электролиты),
полностью
диссоциируют
в растворе
многие соли
неорганических
кислот, некоторые
кислоты и основания.
Неполная
электролитическая
диссоциация
наблюдается
для солей, катионы
которых склонны
к образованию
ковалентных
связей с анионами,
например соли
Ag, Cd, Zn. Некоторые
многоосновные
кислоты, например
H2SO4, полностью
диссоциируют
лишь в отношении
отщепления
одного иона
Н+, а дальнейшая
диссоциация
![]() затруднена.
Разбавленные
растворы слабых
электролитов
по своим свойствам
близки к идеальным
растворам, для
них в формуле
(1) коэффициент
активности
можно считать
равными 1. Тогда
формула (1) переходит
в закон разведения
Оствальда:
затруднена.
Разбавленные
растворы слабых
электролитов
по своим свойствам
близки к идеальным
растворам, для
них в формуле
(1) коэффициент
активности
можно считать
равными 1. Тогда
формула (1) переходит
в закон разведения
Оствальда:
![]()
в
котором а можно
заменить отношением![]() где
где
![]() и
и
![]() -соответственно
эквивалентная
электропроводность
раствора при
данной концентрации
и при бесконечном
разведении.
В соответствии
с законом Оствальда
с уменьшением
концентрации
раствора степень
диссоциации
а и эквивалентная
электропроводность
возрастают,
причем при
бесконечном
разведении
-соответственно
эквивалентная
электропроводность
раствора при
данной концентрации
и при бесконечном
разведении.
В соответствии
с законом Оствальда
с уменьшением
концентрации
раствора степень
диссоциации
а и эквивалентная
электропроводность
возрастают,
причем при
бесконечном
разведении
![]() и
и
![]() Растворы сильных
электролитов
не являются
идеальными
и для их описания
необходим учет
межионного
взаимодействия
даже в области
предельного
разведения.
При определенных
условиях, например
в растворителях
с малой диэлектрической
проницаемостью,
при низких
температурах
или при образовании
многовалентных
ионов, благодаря
сильному
электростатическому
притяжению
противоположно
заряженных
ионов могут
образовываться
ионные ассоциаты,
простейшими
из которых
являются ионные
пары.
Растворы сильных
электролитов
не являются
идеальными
и для их описания
необходим учет
межионного
взаимодействия
даже в области
предельного
разведения.
При определенных
условиях, например
в растворителях
с малой диэлектрической
проницаемостью,
при низких
температурах
или при образовании
многовалентных
ионов, благодаря
сильному
электростатическому
притяжению
противоположно
заряженных
ионов могут
образовываться
ионные ассоциаты,
простейшими
из которых
являются ионные
пары.
Равновесие между сольватированными ионами и ионными парами характеризуется константой диссоциации, аналогично исходному распаду молекул, или обратной ей величиной - константой ассоциации. В приближении электростатического взаимодействия между ионами константа диссоциации контактных ионных пар, образованных двумя ионами с радиусами r+ и r. и зарядовыми числами z+ и z-, может быть рассчитана по формуле:
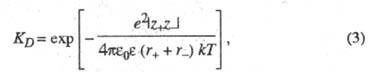
где
е - элементарный
электрический
заряд; k - постоянная
Больцмана;
![]() электрическая
постоянная
(диэлектрическая
проницаемость
вакуума) ;
электрическая
постоянная
(диэлектрическая
проницаемость
вакуума) ;
![]() -
диэлектрическая
проницаемость
растворителя;
Т - абс. температура.
-
диэлектрическая
проницаемость
растворителя;
Т - абс. температура.
Понятие электролитической диссоциации было введено С. Аррениусом в 1887. Электролитическая диссоциация играет важную роль во многих природных и производств, процессах, определяя как свойства растворов электролитов, так и особенности происходящих в них процессов.
Электролиты. Известно, что существуют две основные причины прохождения электрического тока через проводники: либо за счет движения электронов в электрическом поле, либо за счет движения ионов. Электронная проводимость присуща, прежде всего, металлам.
Ионная проводимость присуща многим химическим соединениям, обладающим ионным строением, например солям в твердом или расплавленном состояниях, а также многим водным и неводным растворам. В связи с этим все вещества принято условно делить по их поведению в растворах на две категории: а) вещества, растворы которых обладают ионной проводимостью (электролиты); б) вещества, растворы которых не обладают ионной проводимостью (неэлектролиты). К электролитам относится большинство неорганических кислот, оснований и солей. К неэлектролитам относятся многие органические соединения, например спирты, углеводы. Электролитическая диссоциация. Кроме хорошей электропроводности, растворы электролитов обладают более низкими значениями давления пара растворителя и температуры плавления и более высокими температурами кипения по сравнению с соответствующими значениями для чистого растворителя или для раствора неэлектролита в этом же растворителе. Для объяснения этих свойств шведский ученый С. Аррениус в 1887 г. предложил теорию электролитической диссоциации.Под электролитической диссоциацией понимается распад молекул электролита в растворе с образованием положительно и отрицательно заряженных ионов — катионов и анионов. Процесс диссоциации во всех случаях является обратимым, поэтому при написании уравнений реакции диссоциации необходимо применять знак обратимости «. Различные электролиты, согласно теории Аррениуса, диссоциируют на ионы в различной степени. Полнота распада зависит от природы электролита, его концентрации, природы растворителя, температуры.Степень диссоциации. Одним из важнейших понятий теории электролитической диссоциации Аррениуса является понятие о степени диссоциации.Степенью диссоциации а называется отношение числа молекул, распавшихся на ионы (n'), к общему числу растворенных молекул (п).
Из этого выражения очевидно, что а может изменяться от 0 (диссоциации нет) до 1 (полная диссоциация). Степень диссоциации часто выражают в процентах. Степень диссоциации электролита может быть определена только экспериментальным путем, например по измерению температуры замерзания раствора, по электропроводности раствора и т. д.Сильные и слабые электролиты. В зависимости от степени диссоциации различают электролиты сильные и слабые. Электролиты со степенью диссоциации больше 30% обычно называют сильными, со степенью диссоциации от 3 до 30% — средними, менее 3% — слабыми электролитами.К сильным электролитам относятся почти все соли, некоторые кислоты (НСl, HBr, HI, НNО3, НсlO4, Н2SO4(разб.)) и некоторые основания (LiОН, NaOH, КОН, Са(ОН)2, Sr(OH)2, Ва(ОН)2). К слабым электролитам относится большинство кислот (особенно органических) и оснований.Степень диссоциации как сильных, так и слабых электролитов зависит от концентрации раствора (степень диссоциации тем выше, чем более разбавлен раствор). Константа диссоциации. Более точной характеристикой диссоциации электролита является константа диссоциации, которая от концентрации раствора не зависит.Выражение для константы диссоциации можно получить, если записать уравнение реакции диссоциации электролита АК в общем виде:A K « A- + K+.
Поскольку диссоциация является обратимым равновесным процессом, то к этой реакции применим закон действующих масс, и можно определить константу равновесия как
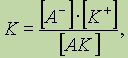
где К — константа диссоциации, которая зависит от температуры и природы электролита и растворителя, но не зависит от концентрации электролита.
Диапазон констант равновесия для разных реакций очень большой — от 10-16 до 1015. Например, высокое значение К для реакции
![]()
означает, что если в раствор, содержащий ионы серебра Ag+, внести металлическую медь, то в момент достижения равновесия концентрация ионов меди [Cu2+] намного больше, чем квадрат концентрации ионов серебра [Ag+]2. Напротив, низкое значение К в реакции
![]()
говорит о том, что к моменту достижения равновесия растворилось ничтожно малое количество иодида серебра AgI.
Обратите особое внимание на форму записи выражений для константы равновесия. Если концентрации некоторых реагентов существенно не изменяются в процессе реакции, то они не записываются в выражение для константы равновесия (такие константы обозначаются К1). Так, для реакции меди с серебром неправильным будет выражение
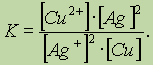
Правильной будет следующая форма записи:
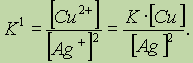
Это объясняется тем, что концентрации металлических меди и серебра введены в константу равновесия. Концентрации меди и серебра определяются их плотностью и не могут быть изменены. Поэтому эти концентрации нет смысла учитывать при расчете константы равновесия.Аналогично объясняются выражения констант равновесия при растворении AgCl и AgI.
Разбавленные растворы электролитов – солей, оснований, кислот в воде – показывают систематические отклонения от свойств идеальных растворов. Эти отклонения связаны с тем, что молекулы электролита в водном растворе распадаются на ионы, и в единице объёма раствора будет содержаться большее число частиц, чем в исходной загрузке соли, кислоты, основания. Для расчета свойств разбавленных растворов электролитов необходимо уравнения законов идеальных растворов исправить, введя в них коэффициент, учитывающий изменение числа частиц в растворе вследствие диссоциации или ассоциации растворенного вещества. Этот коэффициент обозначают i и называют изотоническим коэффициентом. Он показывает отношение числа частиц, образующихся в растворе, к числу частиц в исходной порции вещества. Для электролитов:
АВ = А+ + В-
N(1-б) Nб Nб
б=N1/N
N1 – число образовавшихся ионов или распавшихся молекул
N(1-б) – число нераспавшихся молекул
У Ni = N - Nб +Nб +Nб i= У Ni/N = 1+б
если исходные молекулы распадаются на н новых частиц, то
У Ni = N[1+б(н-1)] i= У Ni/N = 1+б(н-1)
если б=0, то i=1, если б=1, то 1<= i <= н
Для раствора, в котором молекулы растворенных веществ ассоциируют друг с другом:
nA = An
N(1-б) Nб/n
Nб/n – число ассоциированных молекул
N(1-б) – число исходных молекул
У Ni = N - Nб +Nб/n i= У Ni/N = 1+б(1/n -1)
1/n <= i <= 1
С учетом этой поправки законы разбавленных растворов электролитов запишутся:
р = i cRT
∆p/po = i n2/(n1 + i n2), ∆p/po = i n2/n1
i = роп/ррасч = (∆p/p)оп/(∆p/p)расч = ∆T3 оп /∆T3 расч = ∆Tк оп /∆Tк расч
Между активностью a2 сильного электролита в растворе (если формально не учитывать его диссоциацию на ионы) и средней активностью ионов электролита
y±
![]()
Рассмотрим несколько способов определения среднего коэффициента активности электролита y± по равновесным свойствам раствора электролитов.
25. Сильные и слабые электролиты. Константа диссоциации. Закон разбавления Оствальда. Слабые электролиты. Константа диссоциации
Процесс диссоциации слабых электролитов является обратимым и в системе существует динамическое равновесие, которое может быть описано константой равновесия, выраженной через концентрации образующихся ионов и непродиссоциировавших молекул, называемой константой диссоциации. Для некоторого электролита, распадающегося в растворе на ионы в соответствии с уравнением:
АaВb <––> aАx- + bВy+
константа диссоциации выразится следующим соотношением:
![]() (III.21)
(III.21)
Для бинарного (распадающегося на два иона) электролита выражение (III.21) можно переписать в виде (III.21a):
![]() (III.21a)
(III.21a)
Поскольку концентрация каждого иона для бинарного электролита равна произведению степени диссоциации б на общую концентрацию электролита С, выражение (III.21a) в этом случае можно переписать следующим образом:
![]() (III.22)
(III.22)
Для разбавленных растворов можно считать, что (1 – б) = 1. Тогда получаем:
![]() (III.23)
(III.23)
![]() (III.24)
(III.24)
Т.о., степень диссоциации слабого электролита обратно пропорциональна концентрации и прямо пропорциональна разбавлению раствора; выражение (III.24) называют законом разбавления Оствальда. Степень диссоциации слабого электролита можно связать с изотоническим коэффициентом. Будем считать, что из N молекул электролита продиссоциировало n молекул, образовав нn ионов (н – число ионов, на которое диссоциирует молекула). Поскольку изотонический коэффициент показывает, во сколько раз общее число молекул и ионов в растворе больше числа молекул до диссоциации, получаем:
![]() (III.25)
(III.25)
![]() (III.26)
(III.26)
Соотношение (III.26) дает возможность, экспериментально определив изотонический коэффициент раствора, рассчитать степень диссоциации слабого электролита.
Сильные электролиты
Предположение Аррениуса о том, что в растворе сильного электролита также существует динамическое равновесие между молекулами и ионами, как и у слабых электролитов, оказалось ошибочным. Экспериментальные исследования показали, что, во-первых, величина константы диссоциации сильного электролита зависит от концентрации (т.е. к растворам сильных электролитов неприменим закон действующих масс) и, во-вторых, никакими методами не удалось обнаружить в растворах сильных электролитов непродиссоциировавшие молекулы. Это позволило сделать вывод, что сильные электролиты в растворах любых концентраций полностью диссоциируют на ионы и, следовательно, закономерности, полученные для слабых электролитов, не могут применяться к сильным электролитам без соответствующих поправок.
Качественная теория сильных электролитов была разработана П. Дебаем и Г. Хюккелем (1923). Для сильных электролитов, полностью диссоциирующих на ионы, даже при малых концентрациях растворов энергия электростатического взаимодействия между ионами достаточно велика, и пренебречь этим взаимодействием нельзя. Взаимодействие противоположно и одноименно заряженных ионов (соответственно притяжение и отталкивание) приводит к тому, что вблизи каждого иона находятся преимущественно ионы с противоположным зарядом, образующие т.н. ионную атмосферу. Радиус ионной атмосферы сравнительно велик, поэтому ионные атмосферы соседних ионов пересекаются; кроме того, каждый ион окружен дипольными молекулами растворителя – сольватной оболочкой. Т.о., в растворе сильного электролита возникает подобие пространственной структуры, что ограничивает свободу перемещения ионов и приводит к изменению свойств раствора в том же направлении, как действовало бы уменьшение степени диссоциации. Поэтому, определяя степень диссоциации раствора сильного электролита, получают т.н. кажущуюся степень диссоциации, т.е. величину б с поправкой на межионное взаимодействие. Чем выше концентрация раствора, тем сильнее взаимодействие ионов, тем меньше и кажущаяся степень диссоциации сильного электролита.
Количественные расчеты характеристик растворов сильных электролитов осуществляют с помощью понятий активности электролита аэ и активностей катионов и анионов а+ и а- соответственно, которые равны произведению коэффициента активности на концентрацию:
![]() ;
;
![]() ;
;
![]() (III.27)
(III.27)
Для бинарного электролита средняя активность электролита связана с активностями ионов соотношением (III.28); подобным же образом связан средний коэффициент активности с ионными:
![]() (III.28)
(III.28)
![]() (III.29)
(III.29)
Дебаем и Хюккелем был разработан метод расчета среднего коэффициента активности сильного электролита. Для бинарного электролита уравнение имеет следующий вид:
![]() (III.30)
(III.30)
Здесь z – заряд иона, для которого рассчитывается коэффициент активности, I – т.н. ионная сила раствора: некоторый параметр, который одновременно учитывает молярную концентрацию и заряд всех имеющихся в растворе ионов. Ионная сила раствора равна полусумме концентраций всех ионов, умноженных на квадрат их заряда:
![]() (III.31)
(III.31)
Теория Дебая – Хюккеля применима только при концентрациях, не превышающих 0.05 моль/л. Для более концентрированных растворов сильных электролитов количественной теории не существует. Оствальда закон разбавления, соотношение, выражающее зависимость эквивалентной электропроводности разбавленного раствора бинарного слабого электролита от концентрации раствора:
![]()
Здесь К — константа диссоциации электролита, с — концентрация, l и lҐ — значения эквивалентной электропроводности соответственно при концентрации с и при бесконечном разбавлении. Соотношение является следствием действующих масс закона и равенства l/lҐ = a, где a — степень диссоциации. Оствальда закон разбавления выведен В. Оствальдом в 1888 и им же подтвержден опытным путём. Экспериментальное установление правильности Оствальда закона разбавления имело большое значение для обоснования теории электролитической диссоциации.
26 Электролитическая диссоциация воды. Ионное произведение воды. Водородный показатель среды. Понятие об индикаторах
Ио́нное произведе́ние воды́ — произведение концентраций ионов водорода Н+ и ионов гидроксида OH− в воде или в водных растворах, константа автопротолиза воды. Вывод значения ионного произведения воды
Вода, хотя и является слабым электролитом, в небольшой степени диссоциирует:
H2O + H2O ↔ H3O+ + OH−илиH2O ↔ H+ + OH−
Равновесие этой реакции сильно смещено влево. Константу диссоциации воды можно вычислить по формуле:
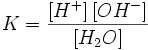
где:
[H+] — концентрация ионов гидроксония (протонов);
[OH−] — концентрация гидроксид-ионов;
[H2O] — концентрация воды (в молекулярной форме) в воде;
Концентрация воды в воде, учитывая её малую степень диссоциации, величина практически постоянная и составляет (1000 г/л)/(18 г/моль) = 55,56 моль/л.
При
25 °C константа
диссоциации
воды равна
1,8Ч10−16моль/л. Уравнение
(1) можно переписать
как:
![]() Обозначим
произведение
K·[H2O] = Kв = 1,8Ч10−16 моль/л·55,56
моль/л = 10−14мольІ/лІ
= [H+]·[OH−] (при 25 °C).
Обозначим
произведение
K·[H2O] = Kв = 1,8Ч10−16 моль/л·55,56
моль/л = 10−14мольІ/лІ
= [H+]·[OH−] (при 25 °C).
Константа Kв, равная произведению концентраций протонов и гидроксид-ионов, называется ионным произведением воды. Она является постоянной не только для чистой воды, но также и для разбавленных водных растворов веществ. C повышением температуры диссоциация воды увеличивается, следовательно, растёт и Kв, при понижении температуры — наоборот. Практическое значение ионного произведения воды
Практическое значение ионного произведения воды велико, так как оно позволяет при известной кислотности (щёлочности) любого раствора (то есть при известной концентрации [H+] или [OH−]) найти соответственно концентрации [OH−] или [H+]. Хотя в большинстве случаев для удобства представления пользуются не абсолютными значениями концентраций, а взятыми с обратными знаком их десятичными логарифмами — соответственно, водородным показателем (pH) и гидроксильным показателем (pOH).
Так
как Kв — константа,
при добавлении
к раствору
кислоты (ионов
H+), концентрация
гидроксид-ионов
OH− будет падать
и наоборот. В
нейтральной
среде [H+] = [OH−] =
![]() моль/л. При
концентрации
[H+] > 10−7 моль/л
(соответственно,
концентрации
[OH−] < 10−7 моль/л)
среда будет
кислой; При
концентрации
[OH−] > 10−7 моль/л
(соответственно,
концентрации
[H+] < 10−7 моль/л) —
щелочной.
моль/л. При
концентрации
[H+] > 10−7 моль/л
(соответственно,
концентрации
[OH−] < 10−7 моль/л)
среда будет
кислой; При
концентрации
[OH−] > 10−7 моль/л
(соответственно,
концентрации
[H+] < 10−7 моль/л) —
щелочной.
Электролитическая диссоциация воды. Водородный показатель рН
Вода представляет собой слабый амфотерный электролит:
Н2О Н+ + ОН-или, более точно:2Н2О Н3О+ + ОН-
Константа
диссоциации
воды при 25оС
равна:
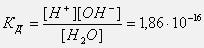 Такое значение
константы
соответствует
диссоциации
одной из ста
миллионов
молекул воды,
поэтому концентрацию
воды можно
считать постоянной
и равной 55,55 моль/л
(плотность воды
1000 г/л, масса 1 л
1000 г, количество
вещества воды
1000г:18г/моль=55,55 моль,
С=55,55 моль: 1 л = 55,55
моль/л). Тогда
Такое значение
константы
соответствует
диссоциации
одной из ста
миллионов
молекул воды,
поэтому концентрацию
воды можно
считать постоянной
и равной 55,55 моль/л
(плотность воды
1000 г/л, масса 1 л
1000 г, количество
вещества воды
1000г:18г/моль=55,55 моль,
С=55,55 моль: 1 л = 55,55
моль/л). Тогда
![]()
Эта величина постоянная при данной температуре (25оС), она называется ионным произведением воды KW:
![]()
Диссоциация воды – процесс эндотермический, поэтому с повышением температуры в соответствии с принципом Ле-Шателье диссоциация усиливается, ионное произведение возрастает и достигает при 100оС значения 10-13.
В чистой воде при 25оС концентрации ионов водорода и гидроксила равны между собой:
[H+] = [OH-] = 10-7 моль/л Растворы, в которых концентрации ионов водорода и гидроксила равны между собой, называются нейтральными. Если к чистой воде прибавить кислоту, концентрация ионов водорда повысится и станет больше, чем 10-7 моль/л, среда станет кислой, при этом концентрация ионов гидроксила мгновенно изменится так, чтобы ионное произведение воды сохранило свое значение 10-14. Тоже самое будет происходить и при добавлении к чистой воде щелочи. Концентрации ионов водорода и гидроксила связаны между собой через ионное произведение, поэтому, зная концентрацию одного из ионов, легко вычислить концентрацию другого. Например, если [H+] = 10-3 моль/л, то [OH-] = KW/[H+] = 10-14/10-3 = 10-11 моль/л, или, если [OH-] = 10-2 моль/л, то [H+] = KW/[OH-] = 10-14/10-2 = 10-12 моль/л. Таким образом, концентрация ионов водорода или гидроксила может служить количественной характеристикой кислотности или щелочности среды.
На практике пользуются не концентрациями ионов водорода или гидроксила, а водородным рН или гидроксильным рОН показателями.Водородный показатель рН равен отрицательному десятичному логарифму концентрации ионов водорода:
рН = - lg[H+]
Гидроксильный показатель рОН равен отрицательному десятичному логарифму концентрации ионов гидроксила:
рОН = - lg[OH-]
Легко показать, прологарифмировав ионное произведение воды, что
рН + рОН = 14
Если рН среды равен 7 - среда нейтральная, если меньше 7 - кислая, причем чем меньше рН, тем выше концентрация ионов водорода. pН больше 7 – среда щелочная, чем больше рН, тем выше концентрация ионов гидроксила. Чистая вода очень плохо проводит электрический ток, но всё же обладает измеримой электропроводностью, которая объясняется небольшой диссоциацией воды на ионы водорода и гидроксид-ионы. По величине электропроводности чистой воды можно определить концентрацию ионов водорода и гидроксид-ионов в воде.
Поскольку степень диссоциации воды очень мала, то концентрация недиссоциированных молекул в воде практически равна общей концентрации воды, поэтому из выражения для константы диссоциации воды получакм, что для воды и разбавленных водных растворов при неизменной температуре произведение концентраций ионов водорода и гидроксид-ионов есть величина постоянная. Эта постоянная величина называется ионным произведением воды.
Растворы, в которых концентрации ионов водорода и гидроксид-ионов одинаковы, называются нейтральными. В кисдых растворах больше ионов водорода, в щелочных - гидроксид-ионов. Но произведение их концентраций всегда постоянно. Это означает, что если известна концентрация ионов водорода в водном растворе, то тем самым и определена и концентрация гидроксид-ионов. Поэтому как степень кислотности, так и степень щёлочности раствора можно количественно охарактеризовать концентрацией ионов водорода:
Кислотность или щёлочность раствора можно выразить более удобным способом: вместо концентрации ионов водорода указывают её десятичный логарифм, взятый с обратным знаком. Последняя величина называется водородным показателем и обозначается рН:. Отсюда ясно, что в нейтральном растворе pH=7; в кислых растворах рН<7 и тем меньше, чем кислее раствор; в щелочных растворах рН>7, и тем больше, чем больше щёлочность раствора.
Для измерения рН существуют различные методы. Приближённо реакцию раствора можно определить с помощью специальных реакторов, называемых индикаторами, окраска которых меняется в зависимости от концентрации ионов водорода. Наиболее распространены метиловый оранжевый, метиловый красный, фенолфталеин и лакмус.
27. Гидролиз солей. Обратимый и необратимый (полный) гидролиз. Роль процессов гидролиза при эксплуатации котельных установок.ъ
В общем случае под гидролизом понимают реакцию разложения вещества водой (от греч. «гидро» - вода, «лизис» - разложение). Гидролизу могут подвергаться белки, жиры, углеводы, эфиры и другие вещества. В неорганической химии чаще всего встречаются с гидролизом солей.
Гидролизом соли называется взаимодействие ионов соли с ионами воды, которое приводит к образованию слабых электролитов. В результате гидролиза солей их водные растворы показывают кислую, щелочную или нейтральную реакцию среды. Как известно, реакция среды зависит от концентрации ионов водорода Н+ или гидроксид-ионов ОН-.
Вода является слабым электролитом и диссоциирует по уравнению
Н2О = Н+ + ОН-.
Появление избытка ионов Н+ или ОН- в растворе объясняется тем, что ионы соли реагируют с ионами воды. Равновесие диссоциации воды смещается вправо, так как при гидролизе солей образуются слабые электролиты:
NH4C1 -> NH4+ + С1- CH3COONa -> СН3СОО~ + Na+
NH4+ + H2O = NH4OH + H+ СН3СОО~ + H2O = CH3COOH + OH-
В зависимости от природы соли в растворе накапливаются либо ионы Н+, либо ОН-, которые и определяют реакцию среды.
Гидролиз соли - это реакция, обратная реакции нейтрализации. Поэтому каждую соль можно представить себе как соединение, образованное основанием и кислотой. Кислоты и основания бывают сильными или слабыми электролитами. В зависимости от силы исходной кислоты и исходного основания различают четыре типа солей :
• образованные сильным основанием и слабой кислотой;
• образованные слабым основанием и сильной кислотой;
• образованные слабым основанием и слабой кислотой;
• образованные сильным основанием и сильной кислотой.
Соли, образованные сильным основанием и слабой кислотой
В водном растворе цианида калия соль полностью распадается на ионы калия К+ и цианид-ионы CN-. Ионы калия К+ и гидроксид-ионы ОН- могут находиться в растворе одновременно в значительных количествах. Ионы водорода Н+ и цианид-ионы CN- взаимодействуют между собой с образованием циановодородной кислоты. Этот процесс схематически может быть представлен следующим образом:
KCN -> К+ + CN-
Н2О + CN- = ОН- + НCN
В результате гидролиза такой соли в растворе находятся полностью продиссоциированная щелочь и слабо диссоциированная кислота. Эта кислота частично диссоциирует на ионы и возвращает в раствор часть ионов Н+ и CN-. Возникает обратная реакция и устанавливается динамическое химическое равновесие:
К+ + CN- + Н2О = К+ + ОН- + HCN.
Следовательно, реакция между цианидом калия и водой является обратимой и проходит не полностью. Такое явление называется обратимым гидролизом.
В результате того, что в растворе образуется сильный электролит гидроксид калия, концентрация гидроксид-ионов ОН- будет значительно больше концентрации ионов водорода Н+. В растворе соли возникает щелочная среда, т.е. рН > 7. Действительно, эксперимент показывает, что 0,1 М раствор этой соли имеет рН 11,1. Гидролиз цианида калия в сокращенной ионной форме можно представить уравнением
CN- + Н2О = ОН- + HCN.
Подобно раствору KCN, раствор ацетата натрия также имеет щелочную среду, что видно из молекулярного и сокращенного ионного уравнений гидролиза :
CHgCOONa + Н2О = СН3СООН + NaOH; СН3СОО- + Н2О = СН3СООН + ОН-.
Сокращенное ионное уравнение показывает, что гидролиз соли, образованной сильным основанием и слабой кислотой, идет по аниону слабой кислоты и реакция среды становится щелочной.
Соли, образованные слабым основанием и сильной кислотой.
Примером такой соли является йодид аммония NH4I. При растворении этой соли в воде катион аммония связывает гидроксид-ион ОН- воды, а ионы водорода накапливаются в растворе:
NH4I + Н2О = NH4OH + HI; NH4+ + Н2О = NH4OH + H+.
В результате гидролиза данной соли в растворе, образуются слабое основание NH4OH и сильная кислота HI. Йодоводородная кислота является сильным электролитом и в водном растворе полностью распадается на ионы. Концентрация ионов водорода становится значительно больше, чем концентрация гидроксид-ионов, и раствор соли имеет кислую среду, т.е рН 7.
Такой же процесс происходит и в случае растворения хлорида аммония NH4C1 в воде:
NH4C1 + Н2О = NH4OH + HC1 или NH4+ + Н2О = NH4OH + H+.
Таким образом, гидролиз соли, образованной слабым основанием и сильной кислотой, идет по катиону слабого основания и реакция среды становится кислой.
Соли, образованные слабым основанием и слабой кислотой
В случае гидролиза солей, образованных слабым основанием и слабой кислотой, оба иона ОН- и Н+ воды связываются. Образуются слабая кислота и слабое основание. CH3COONH4 -> СН3СОО- + NH4+
СН3СОО- + NH4+ +H2O = CH3COOH + СН3СОО- + NH4+
Гидролиз соли идет одновременно и по катиону, и по аниону. В зависимости от константы диссоциации продуктов гидролиза (кислоты и основания) реакция среды растворов таких солей может быть слабокислой, слабощелочной или нейтральной. Например, реакция среды в случае гидролиза ацетата аммония CH3COONH4 — нейтральная, поскольку константы диссоциации СН3СООН и NH4OH равны. В случае же гидролиза соли цианида аммония NH4CN реакция среды слабощелочная.
Таким образом, гидролиз соли, образованной слабым основанием и слабой кислотой, идет одновременно и по катиону, и по аниону. Реакция среды зависит от констант диссоциации продуктов гидролиза.
Соли, образованные сильным основанием и сильной кислотой
Соли этого типа гидролизу не подвергаются, потому что катионы и анионы этих солей не связываются с ионами Н+ и ОН- воды и в растворе не образуются молекулы слабых электролитов. Поскольку связывания ионов воды не происходит, реакция среды растворов этих солей остается нейтральной. Рассмотрим это на примере раствора хлорида натрия. Взаимодействие этой соли с водой можно представить уравнениями
NaCl + Н2О = NaOH + HC1 или Na++ С1- + Н2О = Na+ + ОН- + Н+ + С1-.
Производя сокращения в ионном уравнении, получаем Н2О = Н+ + ОН. Отсюда видно, что ионы соли не участвуют в реакций и среда остается нейтральной.
Следовательно, соли, образованные сильной кислотой и сильным основанием, при растворении в воде гидролизу не подвергаются, а реакция среды остается нейтральной.
Ранее мы рассмотрели гидролиз солей, образованных одноосновными кислотами и однокислотными основаниями. Продуктами гидролиза таких солей являются кислоты и основания.
Если соль образована слабой многоосновной кислотой или слабым многокислотным основанием, то гидролиз данной соли может протекать ступенчато. Число ступеней гидролиза зависит от основности слабой кислоты и кислотности слабого основания.
Рассмотрим гидролиз соли, образованной слабой многоосновной кислотой и сильным основанием. В водном растворе этих солей на первой ступени гидролиза образуется кислая соль вместо кислоты и сильное основание. Ступенчато гидролизуются соли K2Si03, Na2SO3, Na2S, Na3PO4 и др. Например, гидролиз Na2CO3 может быть изображен в виде уравнений.
Первая ступень: Na2CO3 + Н2О = NaHCO3 + NaOH; С032- + Н20 = HCO3- + ОН-
Продуктами первой ступени гидролиза является кислая соль гидрокарбонат натрия NaHCO3 и гидроксид натрия NaOH.
Вторая ступень:
NaHCO3 + Н2О = Н2СО3 + NaOH;
HCO3- + Н2О = Н2СО3 + ОН-.
Продуктами второй ступени гидролиза карбоната натрия Na2CO3 являются гидроксид натрия и слабая угольная кислота Н2СО3. Гидролиз по второй ступени протекает в значительно меньшей степени, чем по первой ступени. Среда раствора соли карбоната натрия Na2CO3 - щелочная (рН > 7), так как в растворе увеличивается концентрация гидроксид-ионов ОН-.
Гидролиз солей трехосновных слабых кислот протекает по трем ступеням. В качестве примера приведем уравнения гидролиза фосфата натрия.
Первая ступень:
Na3PO4 + Н2О = Na2HPO4 + NaOH;
PO43- +Н2О = HPO42- +NaOH.
Вторая ступень:
Na2HPO4 + H2O = NaH2PO4 + NaOH;
НРО42- +Н20 = Н2РО4- + ОН-.
Третья ступень:
NaH2PO4 + Н20 = Н3РО4 + NaOH;
H2PO4- + H2O = Na3PO4 + ОН-.
Гидролиз по первой ступени происходит в значительно большей степени, чем по второй. По третьей ступени гидролиз фосфата натрия практически не идет.
Рассмотрим гидролиз соли, образованной слабым многокислотным основанием и сильной кислотой. В водных растворах таких солей на первой ступени образуется основная соль вместо основания и сильная кислота. Ступенчатому гидролизу подвергаются соли : MgSO4, FeCl3, FeCl2, ZnCl2 и др. Например, гидролиз хлорида цинка ZnCl2 протекает по двум ступеням.
Первая ступень: ZnCl2+ H2O = ZnOHCl + НС1;
Вторая ступень: ZnOHCl+ H2O = Zn(OH)2 + HC1;
Гидролиз соли идет по катиону, так как соль образована слабым основанием Zn(OH)2 и сильной кислотой НС1. Катионы цинка Zn2+ связывают гидроксид-ионы ОН- воды. На первой ступени образуется основная соль ZnOHCl и сильная кислота НС1. На второй ступени образуется слабое основание Zn(OH)2 и тоже сильная хлороводородная кислота. Гидролиз по первой ступени протекает значительно больше, чем по второй. В растворе увеличивается концентрация ионов водорода Н+ и реакция среды будет кислая (рН <7).
Степень гидролиза. Смещение равновесия гидролиза.
Для большинства солей процесс гидролиза обратим. В состоянии равновесия только часть растворенной соли гидролизуется. Количественно гидролиз характеризуется степенью гидролиза h, которую выражают в долях единицы или в процентах.
Степень гидролиза (п) измеряется отношением количества гидролизованного вещества к общему количеству растворенного вещества: h = nr\no
где пr - количество гидролизованной соли, моль; п0 - общее количество растворенной соли, моль.
Например, если из каждых 3 моль соли, растворенной в воде, 0,015 моль подвергается гидролизу, то степень гидролиза равна 0,015/3 = 0,005, или 0,005 • 100°/о=0,5%.Степень гидролиза соли зависит от природы соли, концентрации раствора соли и температуры.
Для солей, образованных катионом сильного основания и анионом слабой кислоты, степень гидролиза зависит от аниона кислоты, который входит в состав этой соли. Например, цианид натрия NaCN и ацетат натрия CH3COONa - это соли, образованные слабой кислотой и сильным основанием. Обе соли подвергаются гидролизу в разной степени, причем гидролиз в растворе NaCN происходит полнее, чем в растворе CH3COONa. Это объясняется тем, что константа диссоциации циановодородной кислоты HCN меньше, чем константа диссоциации уксусной кислоты СН3СООН. Вследствие этого процесс связывания ионов водорода в молекулу HCN идет полнее. Значит, чем слабее кислота, тем сильнее подвергаются гидролизу ее соли. Для соли, образованной сильной кислотой и слабым основанием, степень гидролиза зависит от катиона основания: чем слабее основание, тем сильнее гидролизуется соль.
Особенно глубоко протекает гидролиз солей, образованных слабой кислотой и слабым основанием. Примером этому может служить гидролиз ацетата алюминия, протекающий до основных солей - ацетатов гидроксо- и дигид-роксоалюминия:
А1(СН3СОО)3+ Н2О = А1(ОН)(СН3СОО)2+ СН3СООН;
А1(ОН)СН3СОО)2+ Н2О = А1(0Н)2 (СН3СОО) + СН3СООН.
Рассмотрим для данного случая отдельно гидролиз по катиону и по аниону. Эти процессы выражаются уравнениями
А13+ + Н2О = А1(0Н)2+ + Н+; СН3СОО- + Н2О = СН3СООН + ОН-.
При гидролизе по катиону образуются ионы Н+, а при гидролизе по аниону - ионы ОН-. Эти ионы соединяются в молекулы воды и не могут сосуществовать в значительных концентрациях. Поэтому гидролиз по катиону и гидролиз по аниону в данном случае усиливают друг друга, что приводит к смещению обоих равновесий вправо.
При гидролизе солей устанавливается динамическое химическое равновесие. Это означает, что ни прямая, ни обратная реакции в состоянии равновесия не прекращаются, а идут с одинаковыми скоростями. Изменяя концентрацию одного из реагирующих веществ, можно смещать равновесие влево или вправо. При разбавлении раствора гидролиз соли увеличивается, так как при добавлении воды, согласно принципу смещения равновесия, последнее смещается вправо. Для того чтобы установилось новое равновесие, некоторое количество соли должно гидролизоваться. Например, при разведении водой при 25 °С 1 0,1М раствора карбоната натрия до 0,01М раствора степень гидролиза соли увеличивается от 2,9 % до 11,3 %. Смещать равновесие гидролиза соли можно путем непосредственного изменения концентрации продуктов гидролиза. Если продуктом гидролиза является кислота, то добавление в раствор соли кислоты сдвигает равновесие влево, т.е. в сторону увеличения концентрации негидролизованной соли. В этом случае гидролиз уменьшается. Если же из раствора гидролизованной соли удалять образовавшуюся кислоту, то степень гидролиза увеличивается. Таким образом можно регулировать гидролиз раствора соли ; хлорида железа (III), который сопровождается образованием осадка основных солей :
FeCl3 + Н2О = Fe(OH)Cl2+HC1;
Fe3+ + Н2О = Fe(OH)2+ + H+.
С целью подавления гидролиза раствор подкисляют небольшим количеством сильной кислоты НС1 и равновесие смещается в сторону исходной соли. Этот метод подавления гидролиза используется в лабораторной практике при приготовлении растворов легко гидролизующихся солей.
В некоторых случаях гидролиз надо увеличить, например, обнаружение ионов висмута Bi3+ осуществляют, используя реакцию гидролиза, при которой образуется белый осадок хлорида висмутила ВiOС1:
BiCl3 + 2Н2О = Bi(OH)2Cl + 2НС1;
Bi(OH)2Cl = BiOCl + H2O.
Повышение температуры раствора усиливает гидролиз соли. Это объясняется тем, что при повышении температуры диссоциация воды возрастает, в то время как степень диссоциации кислоты или основания мало изменяется. Так, при разбавлении водой и нагревании раствора ацетата железа (III) происходит гидролиз данной соли. Это используется для качественного обнаружения ацетат-ионов:
Fe (СН3СОО)3 + 2Н2О = Fe(OH)2 СН3СОО + 2СН3СООН.
Таким образом, влияние различных факторов на смещение равновесия гидролиза используется в аналитической химии для обнаружения отдельных ионов, регулирования кислотности и щелочности анализируемых растворов, разделения ионов при систематическом качественном анализе. Процесс гидролиза солей оказывает влияние на проведение количественного анализа, что используется при определении слабых кислот с помощью сильных оснований.
Необратимый, или полный, гидролиз
Гидролиз солей, в результате которого образуются малорастворимые или газообразные продукты, удаляющиеся из сферы реакции, является необратимым. Например, при гидролизе сульфида алюминия A12S3 выделяется газ H2S и образуется осадок А1(ОН)3. В результате соль A12S3 в водных растворах существовать не может:
A12S3 + 6Н2О = 2А1(ОН)3 + 3H2S
Такое явление наблюдается в результате обмерной реакции между водными растворами некоторых солей, когда одна из двух получающихся солей сразу подвергается необратимому гидролизу с образованием соответствующего нерастворимого основания и слабой летучей кислоты:
2СгС13 + 3Na2S = Cr2S3 + 6NaCl;
Cr2S3 + 6Н2О = 2Cr(OH)3 + 3H2.
Суммируя jith два уравнения, получаем
2CrCl3 + 3Na2S + 6H2O = 2Cr(OH)3 + 6NaCl+ 3H2S;
2Cr3+ + 3S2- + 6H2O = 2Cr(OH)3+ 3H2S.
В водных растворах не могут существовать карбонаты хрома и железа, силикат аммония, так как сразу образуются продукты их гидролиза :
2FeCl3 + 3Na2CO3 + 3H2O = 2Fe(OH)3+ 3CO2+ 6NaCl;
Процесс гидролиза применяют для получения ценных веществ из древесины, жиров, эфиров. Особенно важную роль гидролиз играет в жизнедеятельности организмов. При гидролизе аденозинтрифосфата (АТФ) высвобождается энергия, необходимая для жизнедеятельности организмов.
Без ферментативного гидролиза не могли бы усваиваться белки, жиры, полисахариды, так как всасываться в кишечнике способны относительно небольшие молекулы. Например, усвоение дисахаридов и полисахаридов становится возможным лишь после их полного гидролиза ферментами до моносахаридов.
28. Растворимость веществ. Произведение растворимости. Механизм накипеообразования
Растворимость вещества — способность образовывать с другим веществом однородную, термодинамически устойчивую систему переменного состава, состоящую из двух или большего числа компонентов. Такие системы возникают при взаимодействии газов с жидкостями, жидкостей с жидкостями и т.д. (см. Растворы).Соотношение компонентов может быть либо произвольным, либо ограниченным некоторыми пределами. В последнем случае Р. называют ограниченной. Мерой Р. вещества при данных условиях служит концентрация его насыщенного раствора. Р. различных веществ в определённом растворителе зависит от внешних условий, прежде всего — от температуры и давления. Давление наиболее сильно сказывается на Р. газов. Изменение внешних условий влияет на Р. в соответствии с принципом смещения равновесий (см. Ле Шателье — Брауна принцип).Для наиболее важных растворителей составлены таблицы Р. различных веществ в зависимости от внешних условий или только для стандартных условий.
Произведение растворимости — произведение концентраций ионов в насыщенном растворе малорастворимого сильного электролита. Показатели степени для концентраций, входящих в П. р., равны коэффициенту при соответствующем ионе в уравнении диссоциации электролита. Для неидеальных растворов концентрации должны быть заменены на активности и полученное произведение называется произведением активностей. При данной температуре и в данном растворителе П. р. для каждого электролита есть характерная постоянная величина.
Постоянство П. р. выводится из действующих масс закона и представляет собой частную форму этого закона в приложении к равновесию твёрдый электролит Ы его насыщенный раствор. При этом предполагается, что в растворе электролит находится в полностью диссоциированной форме. П. р. наиболее точно измеряется методом эдс. Часто для измерения П. р. используют также определение растворимости по электропроводности насыщенных растворов. Для многих соединений П. р. установлено с достаточной для практических целей точностью. В таблицах П. р. обычно приводятся при температуре 25 °С (иногда при 18 °С).Из правила постоянства П. р. следует, что если произведение концентраций ионов в растворе превышает величину П. р., то выпадает осадок; в противном случае осадок не образуется. Это следствие позволяет регулировать содержание ионов в растворе при использовании процессов осаждения, растворения, а также высаливания, имеющих большое значение в аналитической химии и химической технологии. Так, при увеличении концентрации одного из ионов путём введения в раствор нового электролита с одноимённым катионом или анионом концентрация др. иона понижается за счёт выпадения части труднорастворимого электролита в осадок. Понижение растворимости происходит обычно лишь до некоторого минимального значения, после чего может наблюдаться вновь повышение растворимости из-за образования комплексных ионов или увеличения ионной силы раствора. Повышения растворимости можно достигнуть, связывая один из ионов в растворе, так что образуется др. ион, который не даёт малорастворимого соединения. Например, для перевода в раствор осадка СаСО3 ион связывают с помощью иона Н+ в слабо диссоциированный ион : ; концентрация ионов при этом уменьшается и осадок растворяется до тех пор, пока не будет достигнуто П. р.
Сероводородный метод анализа, метод химического качественного анализа смеси ионов металлов (главным образом катионов) в водных растворах. Метод основан на неодинаковой растворимости хлоридов, гидроокисей, карбонатов и сульфидов металлов.
С. м. а. предполагает классификацию ионов металлов, представленную в таблице. Существуют и другие классификации ионов металлов. Применяя т. н. групповые реагенты — осадители (HCl, H2S, (NH4)2S, (NH4)2CO3), последовательно разделяют сложную по составу смесь ионов металлов на пять аналитических групп.Ход систематического анализа следующий: добавлением HCl выделяют ионы V группы. Из фильтрата (pH около 3) осаждают катионы IV группы пропусканием H2S. Затем действием избытка (NH4)2S переводят в осадок катионы III группы. Оставшуюся в растворе смесь катионов II и I групп разделяют прибавлением раствора (NH4)2CO3. После этого каждую группу катионов разделяют на подгруппы и обнаруживают ионы химическими реакциями.С. м. а. применяется для предварительной идентификации неизвестного по составу вещества с целью выбора наиболее рационального пути его количественного анализа. Количественный С. м. а. иногда используется при анализе сложных по составу материалов.
Накипеобразование на поверхностях нагрева. В процессе работы котла в котловой воде протекают различные физико-химические процессы, обусловливающие разрушение одних соединений и образование других. Это приводит к возникновению веществ с различной степенью растворимости. Труднорастворимые вещества выделяются из воды в виде осадка, образующего при определенных условиях накипь или шлам.
Накипью называют плотные отложения, возникающие на поверхности нагрева. К шламу относятся выпадающие вещества в виде подвижного осадка, которые могут также образовывать вторичную накипь, прикипая к поверхности труб.
Образование осадка в виде накипи или шлама происходит при наличии пересыщенного раствора, т. е. высокой концентрации солей. Испарение котловой воды, подача питательной и добавочной воды с более высокой минерализацией создают благоприятные условия для этого процесса. Произведение концентраций находящихся в растворе ионов труднорастворимого вещества называется произведением растворимости, т.е.
ПР = СКТСАН где СКТ,САН— концентрация соответственно катиона и аниона труднорастворимого соединения. Произведение концентраций при данной температуре является постоянной величиной и, если СКТСАН > ПР, происходит выпадение осадка (твердой фазы). Образующиеся в толще воды кристаллические частицы осаждаются на поверхности нагрева в виде слоя накипи или остаются во взвешенном состоянии как подвижный шлам. Накипь может появиться в результате увеличения концентрации одного из ионов, образующих труднорастворимые соединения, что является следствием химических процессов.Таким образом, низкое содержание Са в воде еще не означает, что не будет кальциевых отложений.Наибольшее влияние на процесс накипеобразования оказывают катионы Са2+ и Mg2+ и анионы С2-3, ОН-, SO2-4, SiO2-3. Определенные сочетания этих катионов и анионов в виде солей представляют собой труднорастворимые вещества. Накипеобразующими соединениями, например, являются: карбонат кальция и магния (СаСО3, MgCO3), гидрат магния (Mg(OH)2), сульфат кальция (CaSO4), силикаты кальция и магния СаSiO3, MgSiO3).
Карбонат кальция образуется в результате нагрева из бикарбоната:
Са(НСО3)2→СаСО3 +H2O+СО2.
Повышение концентрации в воде углекислоты СО2 может смещать равновесие реакции влево, т. е. ведет к образованию бикарбоната. Однако для котловой воды, где идет процесс кипения и СО2 удаляется, наиболее характерен переход Са(НСО3)2 в карбонат СаСО3.
Аналогичная реакция идет и с бикарбонатом магния при нагревании: Mg(HCO3)2 → MgCO3 + Н2О + СО2.
При нагревании воды с высокой щелочностью происходит гидролиз карбоната магния с образованием труднорастворимого соединения гидроокиси магния: MgCO3 + 2Н2О → Mg(OH)2 + H2CO3.
Карбонаты кальция образуют в котле карбонатную накипь. С повышением щелочности воды они осаждаются в грубодисперсном состоянии и входят в состав шлама.
Соединение Mg(OH)2 находится в воде преимущественно в виде шлама и может образовывать вторичную накипь (прикипание осаждающегося шлама).
Силикаты CaSiO3 и MgSiO3 в природной воде находятся в коллоидальной форме в небольшом количестве. Однако в случае образования силикатной накипи на поверхности нагрева слой загрязнения становится прочным, трудноудаляемым.
Одной из причин образования насыщенных растворов и выпадения осадка является понижение растворимости некоторых соединений при повышении температуры воды. Такие соединения имеют отрицательный коэффициент растворимости. К ним относятся СаСО3, CaSO4, Mg(OH)2, CaSiO4, MgSiO3.
Вторичную накипь могут образовывать продукты коррозии металла, заносимые в котел с питательной водой.
29 Окислительно-восстановительные реакции (ОВР), их классификация. Важнейшие окислители и восстановители. Составление уравнений ОВР по методу полуреакций. Влияние среды на протекание ОВР
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ РЕАКЦИИ (ОВР)
ОВР – реакции, в которых изменяются степени окисления элементов, то есть электроны переходят от одного атома или вещества (восстановителя) к другому (окислителю).
А степень окисления – это заряд, который имел бы атом, если бы все образованные им полярные связи стали ионными. Если связи действительно ионные, то с.о. совпадает с зарядом элементарного иона, например, K+F-. Если не все связи ионные, то степень окисления – это условное понятие, не имеющее строгого смысла, но полезное.
Например, реакция С + О2 = СО2 считается ОВР, т.к. в простых веществах степени окисления нулевые, а в СО2 углероду приписывается с.о. +4, а кислороду –2, хотя ионов С+4 и О-2 там нет. Считается, что при сгорании угля электроны переходят от атомов С к атомам О, но это трудно доказать экспериментально. Это пока условность.
Но во многих случаях удается провести ОВР так, что восстановитель и окислитель разделены в пространстве, и заряды передаются через проводники. Тогда можно экспериментально зарегистрировать перенос заряда (токи, напряжения) и определить, сколько именно электронов передается. ОВР становится реальным, а не условным, понятием. Такие процессы изучает ЭЛЕКТРОХИМИЯ. Подробно – в курсе ФХ, а сейчас очень кратко. Ключевое понятие электрохимии –
ЭЛЕКТРОДНЫЙ ПОТЕНЦИАЛ
Электрод – это электронный проводник (металл или полупроводник, твердый или жидкий), находящийся в контакте с электролитом, т.е. ионным проводником (раствором, расплавом или твердым). При этом на границе раздела фаз возникает скачок электрического потенциала – электродный потенциал. Почему?
1) Ионы металла могут в некотором количестве перейти в раствор, оставив электроны в металлической фазе. СХЕМА. Этому способствуют полярные молекулы растворителя, например, воды: М(тв.) + mH2O(ж.) = [M(OH2)m]n+(ж.) + ne-(тв.). В данном примере электрод заряжается отрицательно и притягивает к себе катионы, так что они могут возвращаться обратно, и наступает равновесие. Это самопроизвольный процесс хотя бы потому, что ведет к росту энтропии.
2) Ионы из электролита, как одноименные с материалом электрода, так и посторонние, могут прилипать (адсорбироваться) на поверхности металла, сообщая ему заряд и потенциал, как положительный, так и отрицательный.Абсолютное значение потенциала j невозможно измерить: если к электроизмерительному прибору (вольтметру, потенциометру) присоединить один электрод - прибор ничего не покажет, т.к. цепь не замкнута, а чтобы замкнуть ее, нужно ввести в электролит второй электрод (СХЕМА), и там возникнет свой электродный потенциал, так что прибор покажет РАЗНОСТЬ потенциалов.
Поэтому договорились: выбрать какой-то электрод за начало отсчета, принять для него j=0, а все остальные отсчитывать от него. Точно так же в механике для расчета потенциальной энергии в поле тяготения (mgh) нужно договориться, от какого уровня отсчитывать высоту h - от уровня стола, пола, земли или моря.
В качестве такого электрода принят нормальный водородный электрод. Это пластинка из платины (покрытая мелкораздробленной “платиновой чернью” для увеличения поверхности), находящаяся в растворе с активностью ионов водорода 1 моль/л (т.е. в 1 н растворе сильной кислоты) и обдуваемая водородом под давлением 1 атм. Там, на трехфазной границе тв.-ж.-газ, устанавливается равновесие 2Н+ (ж) + 2е (тв) = Н2 (г). Платина здесь - инертный электрод, служащий для подвода и отвода электронов, но не входящий в уравнение.Система из двух электродов с разными потенциалами, соединенных электролитом, называется ГАЛЬВАНИЧЕСКИМ ЭЛЕМЕНТОМ. Гальванические элементы применяются как химические источники тока, а также для измерения потенциалов - в аналитических целях и в научных исследованиях (подробнее - в конце темы).
Демонстрация: элемент (+)CuЅр-р Cu SO4ЅЅр-р KCl ЅЅр-р ZnSO4ЅZn (-) (вертикальная черта означает границу раздела электрод-электролит, двойная черта - размытую границу разных электролитов).
Пока не вставлен солевой мостик KCl - цепь не замкнута, стрелка вольтметра стоит на нуле. Я его вставляю - стрелка отклоняется. Видно, что цинковый электрод отрицательный, т.е. служит источником электронов, которые идут по внешней цепи к медному электроду, где восстанавливаются ионы меди. Цинк окисляется, и его ионы переходят в раствор: Zn = Zn2+ +2e, ионы меди восстанавливаются, и медь осаждается на пластинке: Cu2+ +2e = Cu. В результате раствор вблизи цинка приобретает положительный заряд, а вблизи меди - отрицательный, поэтому в электролите катионы идут к меди, анионы - к цинку. Реакцию можно провести и при прямом контакте окислителя с восстановителем, но тогда вместо электроэнергии получим тепло.
Электродное равновесие - это состояние, когда на одном и том же электроде с одинаковыми ненулевыми скоростями идут восстановление и окисление, так что суммарный ток через электрод равен нулю. Если ток не равен нулю, равновесие нарушается, и потенциал отклоняется от равновесного значения. Электродвижущая сила (эдс e) гальванического элемента измеряется в равновесных условиях и равна разности равновесных потенциалов: e = j1 - j2. Но для этого нужно, чтобы сопротивление вольтметра было бесконечно велико. Если оно невелико, то в цепи течет заметный ток, равновесие нарушается, и измеряемое напряжение U < e. Эдс - это работа А по перемещению единичного электрического заряда по всей цепи: А = e*q. Если реагирует моль вещества, и каждая молекула или ион отдает или принимает ne, то q = NAne = nF, где F = eNa = 96500 Кл - число Фарадея - заряд моля электронов. А = enF.
Электрод, на котором идет окисление, называется анодом, а где идет восстановление - катодом. В гальваническом элементе анод - источник электронов, то есть имеет отрицательный знак, катод - положительный, а при электролизе все наоборот.
ФАКТОРЫ, ВЛИЯЮЩИЕ НА ЭЛЕКТРОДНЫЙ ПОТЕНЦИАЛ
1. Важнейший, конечно, это природа электрода и электролита (включая природу растворителя). Мы далее будем подробно разбирать окислительно-восстановительные свойства разных веществ, а пока - лишь некоторые соображения на простейшем примере равновесия металла с раствором его соли:
М(тв.) + m H2O(ж.) = [M(OH2)m]n+(ж.) + ne-(тв.).
На первый взгляд может показаться, что этот процесс аналогичен процессу ионизации атома: М(г.) = Мn+(г.) + ne-(г.). Энергия, необходимая для отрыва электрона от атома, называется потенциалом ионизации I (а тут - сумма n потенциалов ионизации). Чем ниже I и чем ниже j, тем легче металл отдает электроны, тем более сильным восстановителем он является. Но все же потенциал ионизации и электродный потенциал - это разные величины, они соответствуют разным процессам. В первом случае ион образуется из обособленного (газообразного) атома, а во втором - атомы связаны в твердое тело, и зачастую весьма прочно. Если бы железо или вольфрам состояли из несвязанных атомов, они бы не были такими твердыми и тугоплавкими! Для получения свободных атомов из твердого металла нужно затратить энергию атомизации. Чем она больше, тем менее активен металл. Ион тоже в первом случае - газообразный, во втором - сольватированный. При сольватации выделяется большая энергия, поэтому с увеличениемЅDсольв.НЅ восстановительные свойства усиливаются. Наконец, и электроны в первом случае газообразные, а во втором - связаны в металле, и их энергии отличаются на работу выхода электрона. Таким образом, электродные потенциалы металлов зависят не только от атомных свойств, но и от прочности связи атомов и электронов в простом веществе, от энергии сольватации. Если электрод инертный (не расходуется и не образуется, как Pt в водородном электроде), то его природа не влияет на равновесный потенциал, но влияет на скорость установления равновесия. Здесь электрод - катализатор. Поэтому часто вместо термина “электродный потенциал” употребляют “окислительно-восстановительный потенциал” или “редокс-потенциал”.
2. Концентрации или парциальные давления окисленной и восстановленной форм.
Качественно ясно по принципу Ле Шателье: чем выше концентрация окисленной формы и чем ниже концентрация восстановленной, тем сильнее эта система притягивает электроны, т.е. тем выше j. Количественную зависимость дает уравнение В. Нернста (без вывода):
j = j0 + (RT/nF) ln([Ox]/[Red]), где
R и T - газовая постоянная и абс. температура, n - число электронов, передаваемых в электродной реакции, [Ox] - концентрация (точнее, активность) или давление окисленной формы в степени, соответствующей коэффициенту в уравнении, причем здесь учитывается не только сам окислитель, но вообще все реагенты, стоящие в одной стороне уравнения с окислителем, а [Red] - то же для восстановленной формы, j0 - стандартный электродный потенциал. Как всегда, если вещество составляет отдельную твердую или жидкую фазу, его активность по определению равна единице и не пишется; если вещество растворено, то подставляется его молярная концентрация, а стандартным состоянием является 1 М раствор; если вещество плохорастворимое, то стандартное состояние - его насыщенный раствор, то есть опять-таки равновесие с отдельной фазой растворяемого вещества, а если вещество в газовой фазе, то ставится его парциальное давление в атмосферах, а стандартное состояние - 1 атм. Если все реагенты и продукты - в стандартных состояниях, то под логарифмом 1, второе слагаемое =0, и j = j0. Если подставить коэффициент перехода от натуральных к десятичным логарифмам, числовые значения R и F и принять Т = 298 К, то
j = j0 + (0,0591В/n)lg([Ox]/[Red]).
Примеры.
j(Zn2+/Zn) = j0 + (0,0591B/2)lg[Zn2+] - без знаменателя; j(H+/H2) = j0 + (0,0591B/2)lg[H+]2/p(H2).
Для реакции MnO4- +8H+ +5e = Mn2+ + 4H2O j = j0 + (0,0591B/5)lg([MnO4-][H+]8/[Mn2+]). Заметьте, что число электронов здесь подсчитано по закону сохранения заряда (без них суммарный заряд ионов слева +7, справа +2) и не требует представления о том, что в перманганате степень окисления марганца 7. Степень окисления - условное понятие, а 5е в этой реакции определяются объективно.
3. Температура. В уравнении Нернста она влияет в двух местах: ведь и стандартный потенциал сам может зависеть от температуры, поэтому однозначно предсказать вид зависимости сложно, но она, безусловно, есть. В справочниках чаще всего приводят стандартные потенциалы при 298 К.
4. Зависит ли j от способа записи уравнения электродной реакции? Мы знаем, что, если умножить левую и правую части химического уравнения на один и тот же коэффициент b, то соответствующие DН, DS, DG увеличиваются в b раз, а К равновесия возводится в степень b. В уравнении Нернста при этом выражение под логарифмом будет возведено в степень b, а число электронов тоже возрастет в b раз. Если вынести показатель из-под знака логарифма, он сократится - и потенциал останется тем же.
Докажем то же иначе. Убыль энергии Гиббса -DG - это работа химической реакции: -DG = А = nFe. Мы увеличиваем DG и n в одинаковое число раз, e не меняется.Если поменять местами левую и правую части уравнения, то DН, DS, DG меняют знак, а К равновесия превращается в обратную величину. С потенциалом же ничего не происходит. Он характеризует не прямую и не обратную реакцию, а состояние их равновесия. Показания вольтметра не изменятся, если переписать уравнение наоборот. Плюс останется плюсом. Поэтому менять знак потенциала (как в учебнике Я.А. Угая) - это грубая ошибка.
Электродные потенциалы не зависят от формы записи уравнения. Но стандартные электродные потенциалы могут зависеть от формы записи, так как там могут подразумеваться разные стандартные состояния. Если в уравнении стоит Cl2(р-р), значит стандартным состоянием является 1 М раствор, а если Cl2 - то парциальное давление хлора 1 атм. Это разные состояния - разные f. Одно и то же окислительно-восстановительное уравнение можно записать для разных сред, например:
MnO4- +4H+ +3e = MnO2Ї+ 2H2O; j° = 1,69 В;
MnO4- + 2H2O +3e = MnO2Ї + 4 ОН-; j° = 0,60 В.
Почему разные потенциалы? В первом случае под стандартным состоянием понимается состояние с [H+] = 1 моль/л (рН = 0), а во втором - с [ОH-] = 1 моль/л (рОН = 0, а рН = 14). Для любого конкретного рН одно и то же значение j получается из обоих уравнений. Это не разные процессы, а один и тот же, записанный по разным стандартам.
До сих пор речь шла о равновесных потенциалах.
Но если через электрод идет ток, то потенциал отклоняется от равновесного. Это явление называется поляризация электрода. Наиболее очевидная (но не единственная) причина поляризации - изменение концентрации реагентов вблизи электрода. У анода возрастает концентрация окисленной формы и уменьшается концентрация восстановленной, и диффузия не успевает их выравнивать по объему раствора, поэтому, согласно уравнению Нернста, потенциал анода увеличивается по сравнению с тем, который соответствует средним по объему концентрациям. И чем больше плотность тока (сила тока на единицу площади), тем сильнее отклонение потенциала от равновесного. На катоде, где идет восстановление, наоборот, растет концентрация восстановленной формы и падает концентрация окисленной, потенциал катода уменьшается. В гальваническом элементе jк>jА, поэтому напряжение U получается меньше эдс. И чем больший ток мы хотим получить от батарейки, тем меньше ее напряжение. Наоборот, при электролизе jк<jА, поэтому, чтобы процесс шел с большой скоростью, приходится прикладывать напряжение больше равновесного. Посмотрим еще раз на медно-цинковый гальванический элемент. Поскольку концентрации растворов близки к стандартным, его эдс должна соответствовать разности стандартных потенциалов медного и цинкового электродов: 0,34В - (-0,76В) = 1,10 В. Реально получается небольшое отклонение - из-за того, что сопротивление вольтметра не бесконечно, и ток через него все же идет, и из-за неточности концентраций. Да и температура не точно 298 К.
Обязательно ли в гальваническом элементе погружать металл в раствор его соли? Вовсе нет, в источниках тока часто используют другие электролиты. Но если мы погружаем цинк в раствор, где концентрация его ионов равна нулю, то получаем из уравнения Нернста потенциал -Ґ. Смысла в этом нет. Электрод с таким потенциалом мгновенно начинает окисляться, в растворе появляются ионы цинка, их концентрация быстро растет, и потенциал нестабилен. Поэтому для наглядности мы используем заданную и довольно большую концентрацию, которая уже не может сильно измениться.
НАПРАВЛЕНИЕ ОВР
Окислительно-восстановительную реакцию можно (хотя бы мысленно) разложить на две полуреакции - окисление восстановителя и восстановление окислителя. Каждой полуреакции с участием электролита соответствует свое значение электродного потенциала (еще его называют редокс-потенциал). Электроны заряжены отрицательно, поэтому они будут стремиться перейти от системы с меньшим j к системе с большим j. ОВР идет самопроизвольно, если у предполагаемого окислителя j больше, чем у предполагаемого восстановителя. Если же мы пытаемся использовать в роли окислителя систему с меньшим j, то реакция не пойдет. Другой вариант рассуждений. Эдс ОВР равна j окислителя - j восстановителя. А DG = -nFe. Чтобы реакция шла самопроизвольно, нужно DG<0, то есть эдс e >0.
Для ОВР, как для любой реакции, DG° = -RTlnK. Тогда -RTlnK = -nFe°, lnK = nFe°/RT, K = exp(nFe°/RT), где e° = j°ок-ля - j°восст-ля - стандартная эдс реакции.
Пример. Возможна ли какая-нибудь ОВР между металлической медью и водным раствором соли железа (3+)? Выпишем возможные полуреакции и найдем в справочнике соответствующие j°. Обратите внимание, что все уравнения полуреакций записываются в стандартной форме как восстановление окислителя со знаком обратимости. Реально, конечно же, в той системе, где j меньше, будет идти обратный процесс - окисление.
(1) Fe3+ + e = Fe2+; j°1 = 0,77 В;
(2) Fe3+ + 3e = Fe; j°2 = - 0,04 В;
(3) Cu 2+ +e = Cu; j°3 = 0,34 В;
j°2 << j°3 <<j°1. Это значит, что вторая система не может быть окислителем, реакция 2Fe3+ + 3Cu = 2Fe + 3Cu2+ не идет. Точнее, ее константа равновесия ничтожно мала: К = exp(-0,38*6/0,059) » 10-17 при 298К. Но первая система может окислять медь, и реакция 2Fe3+ + Cu = 2Fe2+ + Cu2+ идет. Ее константа равновесия К = exp(+0,43*2/0,059) »106 при 298К. Полуреакция (1) идет в прямом направлении, а полуреакция (3) с меньшим j°- в обратном.
Влияние рН на окислительно-восстановительные свойства и направление ОВР
В водных растворах концентрация ионов водорода меняется в очень широких пределах - от нескольких моль/л в кислых растворах до 10-14 - 10-15 моль/л в щелочных, то есть на 15 порядков. Поэтому, если в ОВР образуются или расходуются ионы водорода или гидроксила, то рН очень сильно влияет на направление таких реакций. Рассмотрим на примере трех вариантов восстановления перманганата.
(1) MnO4- +8H+ +5e = Mn2+ + 4H2O; j01 = 1,51 В;
(2) MnO4- +4H+ +3e = MnO2 + 2H2O; j02 = 1,69 В;
(3) MnO4- + e = MnO42-; j03 = 0,56 В.
j1 = j°1 + (0,0591B/5)lg([MnO4-][H+]8/[Mn2+]) = 1,51В + (0,0591B/5)lg([MnO4-]/[Mn2+]) - (8*0,0591/5)pH. Если принять стандартными все концентрации, кроме рН, то
j1 = 1,51В -0,0946В*рН. При рН=7 это дает 0,85В.
Аналогично j2 = 1,69В - 0,0788рН, при рН=7 получается 1,14В, а при рН=14 = 0,59В.
Постройте на одном графике зависимости потенциалов трех систем от рН. Уравнения линейные, поэтому достаточно вычислить j при двух значениях рН (например, 0 и 14) и соединить прямой (прямую 1 довести только до pH=6, т.к. дальше осаждается Mn(OH)2).
Только у третьей системы потенциал не зависит от рН, а у двух других резко падает с ростом рН, то есть окислительные свойства перманганата сильнее всего выражены в кислой среде, а в щелочной его труднее восстановить и, наоборот, соединения низших степеней окисления легче окислить.
Система 3 является самым слабым окислителем и по потенциалу, и по числу принимаемых электронов. Поэтому третий вариант восстановления реализуется только в очень сильно щелочной среде, где получается j3 > j2, и только при недостатке восстановителя. Если же восстановителя достаточно, то будет идти процесс 2. Процесс 2 по потенциалу предпочтительнее, чем 1, но уступает по числу электронов. Более строгим критерием направления процесса будет DG.
Рассмотрим, например, эти три варианта при окислении иодида до иода: I2Ї +2e = 2I-. Стандартный потенциал этого восстановителя, независимо от рН, 0,54В. (А у другого восстановителя потенциал может тоже зависеть от рН).
(1) 2MnO4- + 16H+ +10 I- = 4Mn2+ + 8H2O + 5 I2Ї; n=10;
(2) 2MnO4- + 8H+ + 6I- = 2MnO2 + 4H2O + 3I2Ї; n=6;
(3) 2MnO4- + 2I- = 2MnO42- + I2Ї; n=2;
Выразим эдс этих трех систем через рН, принимая все прочие концентрации стандартными.
e1 = 1,51 - 0,54 + 0,00591lg[H+]16 = 0,97 - 0,0946pH (B); DG = -nFe; D1G/F = -9,7 + 0,946pH (B);
e2 = 1,69 - 0,54 + (0,0591/6)lg[H+]8 = 1,15 - 0,0788pH (B); D2G/F = -6,9 + 0,473pH (B)
e3 = 0,56 - 0,54 = 0,02(B) независимо от рН; D3G/F = - 0,04 B.
Наиболее выгодна та из реакций, у которой меньше DG в расчете на одно и тоже количество вещества (здесь - на 2 моль перманганата). Если в уравнениях конкурирующих реакций разные коэффициенты, то сначала надо их домножить так, чтобы они стали одинаковыми, а потом уже сравнивать DG. Реакции 1 и 2 равновероятны, если D1G=D2G. Решив это уравнение относительно рН, получаем рН=5,9. При меньших рН D1G<D2G, при больших - наоборот. Аналогично, приравняв D3G=D2G, найдем рН=14,5.
Вывод: если восстановитель в избытке, то в стандартных условиях вариант 1 термодинамически выгоднее в кислой среде (рН<5,9), вариант 2 - в нейтральной и щелочной (5,9<рН<14,5), а вариант 3 - лишь в очень сильно щелочной среде (рН>14,5).
Окислительно-восстановительные реакции (ОВР) (реакции окисления-восстановления) происходят с изменением степени окисления атомов, входящих в состав реагирующих веществ. При окислении веществ степень окисления элементов возрастает, при восстановлении - понижается.
Первоначально окислением называли только реакции веществ с кислородом, восстановлением - отнятие кислорода. С введением в химию электронных представлений понятие окислительно-восстановительных реакций было распространено на реакции, в которых кислород не участвует.
В неорганической химии окислительно-восстановительные реакции (ОВР) формально могут рассматриваться как перемещение электронов от атома одного реагента (восстановителя) к атому другого (окислителя), например:
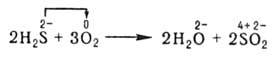
При этом окислитель восстанавливается, а восстановитель - окисляется. При протекании реакций в гальваническом элементе переход электронов осуществляется по проводнику, соединяющему электроды элемента, и изменение энергии Гиббса ДG в данной реакции может быть превращено в полезную работу. В отличие от реакций ионного обмена окислительно-восстановительные реакции (ОВР) в водных растворах протекают, как правило, не мгновенно.
При окислительно-восстановительных реакциях атомы в высшей степени окисления являются только окислителями, в низшей - только восстановителями; атомы в промежуточной степени окисления в зависимости от типа реакции и условий ее протекания могут быть окислителями или восстановителями. Многие окислительно-восстановительные реакции (ОВР) - каталитические По формальным признакам окислительно-восстановительные реакции (ОВР) разделяют на межмолекулярные (например, 2SO2 + O2 → SO3) и внутримолекулярные, например:
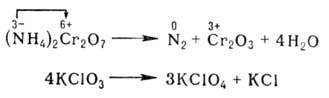
Последняя реакция представляет собой самоокисление-самовосстановление (см. Диспропорционирование).
окислительно-восстановительные реакции (ОВР) часто сопровождаются высоким энерговыделением, поэтому их используют для получения теплоты или электрической энергии. Наиболее энергичные окислительно-восстановительные реакции (ОВР) протекают при взаимодействии восстановителей с окислителями в отсутствие растворителя; в растворах такие реакции могут быть невозможны вследствие окислительно-восстановительного взаимодействия одного или обоих реагентов с растворителем. Так, в водном растворе нельзя непосредственно провести реакцию 2Na + F2 → 2NaF, поскольку натрий и фтор бурно взаимодействуют с водой. На окислительно-восстановительные свойства ионов сильно влияет комплексообразование, например: комплекс [Co2+(CN)6]4-, в отличие от гидратированного иона Со2+, является сильным восстановителем.
В случае окислительно-восстановительных реакций в органической химии использование обобщенной концепции окисления-восстановления и понятия о степени окисления часто малопродуктивно, особенно при незначительно полярности связей между атомами, участвующими в реакции. В органической химии окисление рассматривают обычно как процесс, при котором в результате перехода электронов от органического соединения к окислителю возрастает число (или кратность) кислородсодержащих связей (С — О, N — О, S — О и т.п.) либо уменьшается число водородсодержащих связей (С — Н, N —Н, S —Н и т.п.), например: RCHO → RCOOH; R2CHCHR2 → R2C=CR2. При восстановлении органических соединений в результате приобретения электронов происходят обратные процессы, например: R2CO → R2CH2; RSO2Cl → RSO2H.
Используют также подход, при котором атомам С в молекуле приписывают различные степени окисления в зависимости от числа связей, образованных с элементом более электроотрицательным, чем водород. В этом случае функциональные производные можно расположить в порядке возрастания их степени окисления. Так, насыщенные углеводороды относят к нулевой группе (приблизительная степень окисления — 4), R2C=CR2, ROH, RCl и RNH2 - к первой (- 2), RCCR, R2CO и R2CCl2 - ко второй (0), RCOOH, RCCCl, RCONH2 иRССl3 - к третьей (+2), RCN, CCl4 и СО2 - к четвертой (+4). Тогда окисление - процесс, при котором соединение переходит в более высокую категорию, а восстановление - обратный процесс.
Механизмы окислительно-восстановительных реакций весьма разнообразны; реакции могут протекать как по гетеролитическому, так и по гомолитическому механизму. Во многих случаях начальная стадия реакции - процесс одноэлектронного переноса. Окисление обычно протекает по положениям с наибольшей электронной плотностью, восстановление - по положениям, где электронная плотность минимальна. В органической химии используют широкий ряд восстановителей и окислителей, что позволяет выбрать реагент, обладающий селективностью (т.е. способностью действовать избирательно на определенные функциональные группы), а также получать продукты в требуемой степени окисления. Например, борогид Na восстанавливает кетоны или альдегиды до спиртов, не реагируя с амидами и сложными эфирами; LiAlH4 восстанавливает все эти соединения до спиртов. Среди окислителей высокой селективностью обладают, например, комплекс CrО3 с пиридином, с высоким выходом окисляющий спирты в кетоны, не затрагивая кратные связи С—С, а также SeO2, окисляющий кетоны и альдегиды до б-дикарбонильных соединений.
Селективность окислительно-восстановительных реакций может быть обеспечена и в каталитических процессах; например, в зависимости от катализатора и условий реакций ацетиленовые углеводороды можно селективно гидрировать до этиленовых или насыщенных углеводородов (см. Гидрирование). Электрохимическое восстановление СО2 до СО в водной среде в присутствии никелевого комплекса 1,4,8,11-тетраазациклотетрадекана позволяет проводить желаемый процесс при более низких потенциалах и одновременно подавлять электролиз воды с образованием Н2. Эта реакция имеет ключевое значение для превращения СО2 через СО в разнообразные органические вещества.
Каталитические окислительно-восстановительные реакции (ОВР) играют важную роль в промышленности, например:
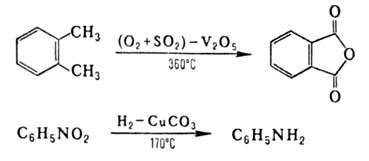
окислительно-восстановительные реакции (ОВР) широко распространены в природе и используются в технике. В основе жизни лежат окислительно-восстановительные реакции (ОВР), происходящие при фотосинтезе, дыхании, транспорте электронов; они же обеспечивают основную часть энергопотребления человечества за счет сжигания органического топлива. Получение металлов, извлечение энергии взрыва основано на окислительно-восстановительных реакциях.
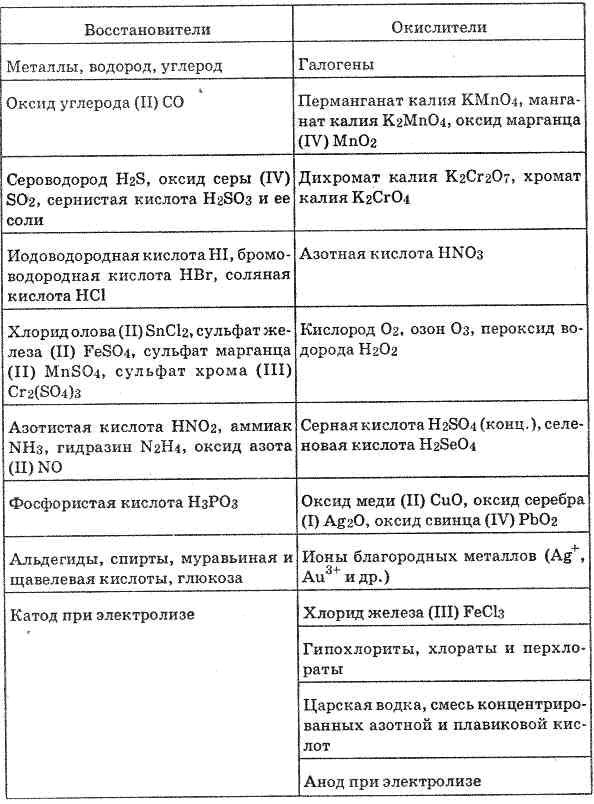
Классификация окислительно-восстановительных реакций
Реакции межмолекулярного окисления-восстановления – это реакции, в которых атом-окислитель и атом-восстановитель принадлежат разным веществам. Эти вещества могут быть как простыми, так и сложными.
4N3-H3 + 3O02 ® 2N02 + 6H2O2-
![]() 2N3-
- 6e ®
N02 2 восстановитель
2N3-
- 6e ®
N02 2 восстановитель
O02 + 4e ® 2O2- 3 окислитель
Реакции внутримолекулярного окисления-восстановления – это реакции, в которых атом-окислитель и атом-восстановитель входят в состав одной и той же молекулы или одного и того же иона.
Например, разложение хлората калия
![]() 2KCl5+O2-3
® 2KCl-
+ 3O02
2KCl5+O2-3
® 2KCl-
+ 3O02
Cl5+ + 6e ® Cl- 2 окислитель
2O2- -4e ® O02 3 восстановитель
Реакции диспропорционирования (самоокисление– самовосстановление) - это реакции, в которых функцию окислителя и восстановителя выполняет один и тот же атом молекулы или иона, находящийся в промежуточной степени окисления. Например:
![]()
![]() 4KCl5+O3
® 3
KCl7+O4
+ KCl-
4KCl5+O3
® 3
KCl7+O4
+ KCl-
Cl5+ - 2e ® Cl7+ 6 3 восстановитель
Cl5+ +6e ® Cl- 2 1 окислитель
Реакции конмутации – реакции внутримолекулярного окисления- восстановления, в результате которых происходит выравнивание степеней окисления атомов одного и того же элемента. Например:
N3-H4N5+O3 = N21+O + 2H2O
Влияние среды на протекание окислительно-восстановительных реакций
Реакции окисления-восстановления могут протекать в различных средах (кислой, нейтральной и щелочной), при этом в зависимости от среды может изменяться характер протекания реакции между одними и теми же веществами. Рассмотрим взаимодействие перманганата калия с сульфитом калия в различных средах.
а) Кислая среда:
2KMn7+O4 + 5K2S4+O3 + 3H2SO4 ® 2Mn2+SO4 + 6K2S6+O4 + 3H2O
![]() Mn7+
+ 5e ® Mn2+ 2
Mn7+
+ 5e ® Mn2+ 2
S4+ - 2e ® S6+ 5
б) Нейтральная среда:
2KMn7+O4 + 3K2S4+O3 + H2O ® 2Mn4+O2 + 3K2S6+O4 + 2KOH
![]() Mn7+
+ 3e ® Mn4+ 2
Mn7+
+ 3e ® Mn4+ 2
S4+ - 2e ® S6+ 3
в) Щелочная среда:
2KMn7+O4 + K2S4+O3 + 2KOH ® K2Mn6+O4 + 2K2S6+O4 +H2O
![]() Mn7+
+ e ® Mn6+ 2
Mn7+
+ e ® Mn6+ 2
S4+ - 2e ® S6+ 1
Схематически это можно представить следующим образом:
Окисленная Восстановленная форма
Форма
![]()
![]() Mn2+
- бесцветный
Mn2+
- бесцветный
![]() Mn7+
® MnО2 -
бурый осадок
Mn7+
® MnО2 -
бурый осадок
![]() MnО42-
- зеленый
MnО42-
- зеленый
Особые случаи составления уравнений окислительно-восстановительных реакций
Рассмотренная методика составления окислительно-восстановительных реакций применима к большинству простых и сложных процессов. Но в некоторых специальных случаях необходимы дополнительные пояснения.
1. Если число электронов, отдаваемое восстановителем, и число электронов, присоединяемое окислителем, имеют общий наибольший делитель, то при нахождении коэффициентов оба числа делят на него. Например, в реакции
HCl7+O4 + 4S4+O2 + 4H2O = 4H2S6+O4 + HCl1-
основными коэффициентами для восстановителя и окислителя будут не 6 и 2, а 4 и 1.
Если число участвующих в реакции электронов нечетно, а в результате получается четное число атомов, то коэффициенты удваиваются. Например, в реакции
2Fe3+Cl3 + 2HJ1- = J20 + 2Fe2+Cl2 + 2HCl
основными коэффициентами будут не 1 и 1, а 2 и 2.
2. Окислитель или восстановитель иногда дополнительно расходуется на связывание получающихся продуктов (солеобразование).Например, в реакции
Cu + 2HNO3 + 6HNO3 = 3Cu(NO3)2 + 2NO + 4H2O
на окисление на связывание на 3 атома восстановителя Сu0 требуется для окисления 2 молекулы окислителя HNO3; кроме того, на образование нитрата меди - трех молекул - требуется еще 6 молекул HNO3 для связывания трех атомов меди. Таким образом, общий расход азотной кислоты: 2 молекулы на окисление плюс 6 молекул на связывание (солеобразование), то есть всего 8 молекул HNO3. И окончательно уравнение примет вид:
3Cu + 8HNO3 = 3Cu(NO3)2 + 2NO + 4H2O.
З. Если в реакции число элементов, изменяющих свою степень окисления, больше двух, то устанавливают общее число электронов, отдаваемых восстановителями, и общее число электронов, присоединяемых окислителями, а в остальном соблюдается общий порядок составления уравнения реакции. Например,
3As3+2S2-3 + 28HN5+O3 + 4H2O ® 6H3As5+O4 + 9H2S6+O4 + 28N2+O
![]()
![]() 2As3+
- 4e ®
2As5+ -28e 3
2As3+
- 4e ®
2As5+ -28e 3
![]() 3S2-
- 24 ®
3S6+
3S2-
- 24 ®
3S6+
N5+ + 3e ® N2+ +3e 28
4. Оба элемента – и окислитель, и восстановитель – находятся в одной и той же молекуле. Это реакция внутримолекулярного окисления-восстановления и реакции диспропорционирования. Для удобства подбора коэффициентов в этом случае иногда можно рассматривать процесс как бы идущим справа налево. Например,
3HN3+O2 ® HN5+O3 + 2 N2+O + H2O
![]() N3+
+ e ®
N2+ 2
N3+
+ e ®
N2+ 2
N3+ - 2e ® N5+ 1
Продукты окислительно-восстановительных реакций устанавливаются опытным путём

Элементы, имеющие высшую степень окисления, могут быть только окислителями.
Элементы, имеющие низшую степень окисления, могут быть только восстановителями.
Элементы, находящиеся в промежуточной степени окисления, могут и окисляться, и
восстанавливаться, то есть проявляют окислительно-восстановительную двойственностьТипичные окислители: металлы, соединения неметаллов в низшей степени окисленияТипичные восстановители: галогены, О2, N2, вещества, содержащие элемент в высшей СО.
Реакции окисления - восстановления могут протекать в различных средах: в кислой, нейтральной и щелочной. В зависимости от среды может изменяться характер протекания реакции между одними и теми же веществами. Среда влияет на изменение степеней окисления. Рассмотрим пример влияния среды на восстановление перманганат- иона MnO4
Классификация ОВР
1. К межмолекулярным относятся реакции, у которых окислитель и восстановитель находятся в разных веществах.
2. К внутримолекулярным относятся реакции, которые протекают с изменением степени окисления атомов в одной и той же молекуле.
3. К реакциям диспропорционирования, или дисмутации (самоокисления-самовосстановления), относятся реакции, сопровождающиеся одновременным увеличением и уменьшением степени окисления атомов одного и того же элемента.
Эти реакции характерны для веществ, содержащих атомы с промежуточной степенью окисления
30. Электрохимические процессы. Гальванический элемент. ЭДС гальванического элемента и его измерение
Гальванический элемент. ЭДС гальванического элемента
Рассмотрим простейший гальванический элемент Даниэля-Якоби, состоящий из двух полуэлементов – цинковой и медной пластин, помещенных в растворы сульфатов цинка и меди соответственно, которые соединены между собой посредством электролитического ключа – например, полоски бумаги, смоченной раствором какого-либо электролита. Схематически данный элемент изображается следующим образом:
Zn / Zn2+ // Cu2+ / Cu
На поверхности каждого из электродов имеет место динамическое равновесие перехода ионов металла из электрода в раствор и обратно, характеризуемое потенциалом ДЭС (зарядом на электроде q). Если соединить медный и цинковый электроды металлическим проводником, немедленно произойдет перераспределение зарядов – электроны начнут перемещаться с электрода с более отрицательным зарядом (в нашем случае – цинкового) на электрод с более положительным зарядом (медный), т.е. в проводнике возникнет электрический ток. Изменение величины заряда каждого из электродов нарушает равновесие – на цинковом электроде начнется процесс перехода ионов из электрода в раствор (окисление металла), на медном – из раствора в электрод (восстановление металла); при этом протекание процесса на одном электроде обусловливает одновременное протекание противоположного процесса на другом:
Zno ––> Zn2+ + 2е-
Сu2+ + 2е- ––> Сuo
Электрод, на котором при работе гальванического элемента протекает процесс окисления, называется анодом, электрод, на котором идет процесс восстановления – катодом. При схематическом изображении гальванических элементов слева записывают анод, справа – катод (стандартный водородный электрод всегда записывают слева). Суммарный окислительно-восстановительный процесс, происходящий в гальваническом элементе, выражается следующим уравнением:
Сu2+ + Zno ––> Сuo + Zn2+
Т.о., гальванический элемент можно определить как прибор для преобразования химической энергии окислительно-восстановительной реакции в электрическую за счет пространственного разделения процессов окисления и восстановления. Работа, которую может совершить электрический ток, вырабатываемый гальваническим элементом, определяется разностью электрических потенциалов между электродами (называемой обычно просто разностью потенциалов) ДЦ и количеством прошедшего по цепи электричества q:
![]() (III.39)
(III.39)
Работа тока гальванического элемента (и, следовательно, разность потенциалов), будет максимальна при его обратимой работе, когда процессы на электродах протекают бесконечно медленно и сила тока в цепи бесконечно мала. Максимальная разность потенциалов, возникающая при обратимой работе гальванического элемента, есть электродвижущая сила (ЭДС) гальванического элемента.
Электрохимические процессы, как и окислительно-восстановительные реакции (ОВР), связаны с изменением степени окисления веществ, участвующих в реакции. Основное отличие ОВР от электрохимических процессов заключается в том, что процессы восстановления и окисления пространственно разделены и перенос электронов может быть зафиксирован как некоторый ток (в гальваническом элементе, при коррозии) или, наоборот, электрохимический процесс может происходить за счет внешнего источника тока (электролиз).
В любом случае для протекания электрохимической реакции необходима электрохимическая цепь, существенными компонентами которой являются электроды и электролит (водный или неводный).
Под электродами обычно понимают или собственно некий проводник или систему, состоящую из проводника, погруженного в раствор электролита. При контакте металлического проводника с раствором электролита на его поверхности возникает некий заряд, за счет переноса электронов, что приводит к возникновению разности электростатических потенциалов между электродом и находящимся с ним в контакте электролитом. Эта разность называется электродным потенциалом.Абсолютную величину электродного потенциала отдельного электрода измерить невозможно, поэтому измеряют всегда разность потенциалов исследуемого электрода и некоторого стандартного электрода сравнения, т.е. составляют электрохимическую цепь. В качестве электродов сравнения для водных сред используют хлорсеребряный или обратимый водородный электрод сравнения. Последний представляет собой платинированную (электрохимическим способом осажденную на платиновую пластину) платину, погруженную в раствор кислоты (серной, соляной) с активностью ионов водорода равной 1, через который продувают водород при давлении 101кПа. В системе устанавливается равновесие
H2(г) H2(Pt) 2H(Pt) 2H+ +2e(Pt)
Потенциал этого равновесия в указанных условиях принят равным нулю при любых температурах.
Табличные значения стандартных электродных потенциалов (Eo) приведены относительно обратимого водородного электрода. Эти значения нормированы на один электрон и их относят к процессу восстановления: Ox + ne =Red
В практических работах в качестве электрода сравнения чаще, чем водородный, используют хлорсеребряный электрод. Хлорсеребряный электрод представляет собой серебряную проволоку, электролитически покрытую AgCl, помещенную в насыщенный раствор KCl.
Стандартные электродные потенциалы металлов и водорода, расположенные в порядке их возрастания, составляют ряд стандартных электродных потенциалов металлов, или электрохимический ряд напряжений металлов. Ряд электродных потенциалов дает полезные знания:
1.Металлы, имеющие значения электродного потенциала меньше, чем у водорода, могут растворяться с выделением водорода в кислотах, анионы которых не являются окислителями.
2.Металлы, имеющие большее, чем у водорода, значение стандартного электродного потенциала могут встречаться в природе в самородном виде.
3.Металлы, имеющие меньшее значение электродного потенциала могут вытеснять металлы с большим значением электродного потенциала из растворов их солей.
4.Металлы, имеющие электродный потенциал меньше, чем потенциал реакции
2H2O + 2e = H2 + 2OH- Eo = –0,83В
в стандартных условиях могут растворяться в воде с выделением водорода.
Под гальваническим элементом понимают единичные ячейки химических источников тока, предназначенных для однократного электрического разряда. Гальванический элемент представляет собой два электрода различной природы и электролит. Максимальная разность потенциалов этих электродов в отсутствие электрического тока называется электродвижущей силой (э.д.с.) гальванического элемента. Э.д.с. может быть рассчитана как разность равновесных потенциалов этих электродов.
Для гальванического элемента, составленного из железного и медного электродов э.д.с. будет равна:
Fe 2+ + 2e =Fe Eo= –0,44
Cu 2+ +2e=Cu Eo=+0,34
э.д.с. =+ 0,34 – ( –0,44) =0,77В,. Электродный потенциал. Уравнение Нернста
ЭДС гальванического элемента E удобно представлять в виде разности некоторых величин, характеризующих каждый из электродов – электродных потенциалов; однако для точного определения этих величин необходима точка отсчета – точно известный электродный потенциал какого-либо электрода. Электродным потенциалом электрода еэ называется ЭДС элемента, составленного из данного электрода и стандартного водородного электрода (см. ниже), электродный потенциал которого принят равным нулю. При этом знак электродного потенциала считают положительным, если в таком гальваническом элементе испытуемый электрод является катодом, и отрицательным, если испытуемый электрод является анодом. Необходимо отметить, что иногда электродный потенциал определяют как "разность потенциалов на границе электрод – раствор", т.е. считают его тождественным потенциалу ДЭС, что не вполне правильно (хотя эти величины взаимосвязаны).
Величина электродного потенциала металлического электрода зависит от температуры и активности (концентрации) иона металла в растворе, в который опущен электрод; математически эта зависимость выражается уравнением Нернста (здесь F – постоянная Фарадея, z – заряд иона):
![]() (III.40)
(III.40)
В уравнении Нернста е° – стандартный электродный потенциал, равный потенциалу электрода при активности иона металла, равной 1 моль/л. Стандартные электродные потенциалы электродов в водных растворах составляют ряд напряжений. Величина е° есть мера способности окисленной формы элемента или иона принимать электроны, т.е. восстанавливаться. Иногда различием между концентрацией и активностью иона в растворе пренебрегают, и в уравнении Нернста под знаком логарифма фигурирует концентрация ионов в растворе. Величина электродного потенциала определяет направление процесса, протекающего на электроде при работе гальванического элемента. На полуэлементе, электродный потенциал которого имеет большее (иногда говорят – более положительное) значение, будет протекать процесс восстановления, т.е. данный электрод будет являться катодом.
Рассмотрим расчёт ЭДС элемента Даниэля-Якоби с помощью уравнения Нернста. ЭДС всегда является положительной величиной и равна разности электродных потенциалов катода и анода:
![]() (III.41)
(III.41)
![]() (III.42)
(III.42)
![]() (III.43)
(III.43)
![]() (III.44)
(III.44)
![]() (III.45)
(III.45)
Как видно из уравнения (III.45), ЭДС элемента Даниэля-Якоби зависит от концентрации (точнее говоря, активности) ионов меди и цинка; при их равных концентрациях ЭДС элемента будет равна разности стандартных электродных потенциалов:
![]() (III.46)
(III.46)
Анализируя уравнение (III.45), можно определить предел необратимой работы гальванического элемента. Поскольку на аноде идет процесс окисления цинка, концентрация ионов цинка при необратимой работе гальванического элемента постоянно увеличивается; концентрация ионов меди, напротив, уменьшается. Отношение концентраций ионов меди и цинка постоянно уменьшается и логарифм этого отношения при [Сu2+] < [Zn2+] становится отрицательным. Т.о., разность потенциалов при необратимой работе гальванического элемента непрерывно уменьшается; при E = 0 (т.е. ек = еа) гальванический элемент не может совершать работу (необратимая работа гальванического элемента может прекратиться также и в результате полного растворения цинкового анода).
Уравнение (III.45) объясняет также и работоспособность т.н. концентрационных цепей – гальванических элементов, состоящих из двух одинаковых металлических электродов, опущенных в растворы соли этого металла с различными активностями а1 > а2. Катодом в этом случае будет являться электрод с большей концентрацией, т.к. стандартные электродные потенциалы обоих электродов равны; для ЭДС концентрационного гальванического элемента получаем:
![]() (III.47)
(III.47)
Единственным результатом работы концентрационного элемента является перенос ионов металла из более концентрированного раствора в менее концентрированный. Т.о., работа электрического тока в концентрационном гальваническом элементе – это работа диффузионного процесса, который проводится обратимо в результате пространственного разделения его на два противоположных по направлению обратимых электродных процесса.
Стандартный (нормальный) водородный электрод. Стандартный электродный потенциал. Таблицы стандартных окислительно-восстановительных потенциалов
В электрохимии стандартный электродный потенциал, обозначаемый Eo, E0, или EO, является мерой индивидуального потенциала обратимого электрода (в равновесии) в стандартном состоянии, которое осуществляется в растворах при эффективной концентрации в 1 моль/кг и в газах при давлении в 1 атмосферу или 100 кПа (килопаскалей). Объёмы чаще всего взяты при 25 °C. Основой для электрохимической ячейки, такой как гальваническая ячейка всегда является окислительно-восстановительная реакция, которая может быть разбита на две полуреакции: окисление на аноде (потеря электрона) и восстановление на катоде (приобретение электрона). Электричество вырабатывается вследствие различия электростатического потенциала двух электродов. Эта разность потенциалов создаётся в результате различий индивидуальных потенциалов двух металлов электродов по отношению к электролиту. Вычисление стандартных электродных потенциалов
Электродный потенциал не может быть получен эмпирически. Потенциал гальванической ячейки вытекает из "пары" электродов. Таким образом, невозможно определить величину для каждого электрода в паре, используя эмпирически полученный потенциал гальванической ячейки. Для этого установлен стандартный водородный электрод, для которого этот потенциал точно определён и равен 0,00 В, и любой электрод, для которого электронный потенциал ещё неизвестен, может быть соотнесён со стандартным водородным электродом с образованием гальванической ячейки - и в этом случае потенциал гальванической ячейки даёт потенциал неизвестного электрода.
Так как электродные потенциалы традиционно определяют как восстановительные потенциалы, знак окисляющегося металлического электрода должен быть изменён на противоположный при подсчёте общего потенциала ячейки. Также нужно иметь в виду, что потенциалы не зависят от количества передаваемых электронов в полуреакциях (даже если оно различно), так как они рассчитаны на 1 моль переданных электронов. Отсюда при расчёте какого-либо электродного потенциала на основании двух других следует проявлять внимательность.
Например:
(ур-е 1) Fe3+ + 3e− --> Fe(тв) -0.036 В
(ур-е 2) Fe2+ + 2e− --> Fe(тв) -0.44 В
Для получения третьего уравнения:
(ур-е 3) Fe3+ + e− --> Fe2+ (+0.77 В)
следует умножить потенциал первого ур-я на 3, перевернуть ур-е 2 (поменять знак) и умножить его потенциал на 2. Сложение этих двух потенциалов даст стандартный потенциал ур-я 3.
Таблица стандартных электродных потенциалов
Основная статья: Таблица стандартных электродных потенциалов
Чем больше стандартные восстановительные потенциалы, тем легче их можно восстановить, другими словами, тем более сильными окислителями они являются. И наоборот: большой отрицательный потенциал означает, что данная форма является сильным восстановителем. Например, F2 имеет 2,87 В, а Li+ имеет -3,05 В, фтор - окислитель, литий - восстановитель. Таким образом, Zn2+, стандартный восстановительный потенциал которого равен -0,76 В, может быть окислен любым другим электродом, стандартный потенциал которого больше -0,76 В. (напр., H+(0 В), Cu2+(0,16 В), F2(2,87 В)) и может быть восстановлен любым электродом, стандартный потенциал которого меньше -0,76 В (напр., H−(-2,23 В), Na+(-2,71 В), Li+(-3,05 В)).В гальванической ячейке, где самопроизвольная окислительно-восстановительная реакция заставляет ячейку производить электрический потенциал, Энергия Гиббса ДGo должна быть отрицательной, в соответствии со следующим уравнением:
ДGoяч = -nFEoяч
где n это количество молей электронов на моль продуктов, а F является постоянной Фарадея, ~96485 Кл/моль. Таким образом применимы следующие правила:
если Eoяч> 0, тогда процесс самопроизвольный (гальваническая ячейка)
если Eoяч< 0, тогда процесс несамопроизвольный (электролитическая ячейка)
Нестандартные условия
Стандартные электродные потенциалы даны при стандартных условиях. Однако, реальные ячейки могут действовать и при нестандартных условиях. При данном стандартном потенциале, потенциал при нестандартных эффективных концентрациях может быть вычислен с использованием уравнения Нернста:
Величины E0 зависят от температуры (кроме стандартного водородного электрода) и обычно относятся к стандартному водородному электроду при этой температуре. Для конденсированных фаз, величины потенциалов также зависят от давления.
Потенциал. Из курса физики известно, что электрический потенциал- работа по перемещению единичного положительного заряда из - данной точки пространства в бесконечность. Каждый электрод обладает каким-то электрическим потенциалом. Абсолютное значение потенциала электрода определить нельзя. Можно лишь сравнивать потенциалы различных электродов друг с другом. Для этого надо два электрода объединить в электрохимическую цепь. Для этого металлические части соединяются проводником, а растворы электролитов, в которые они погружены-стеклянной трубкой, заполненной раствором электролита (обычно хлорида калия). Эту трубку называют электролитическим ключом или солевым мостиком. Она обеспечивает ионную проводимость между растврами. Таким образом возникает замкнутая цепь или гальванический элемент, который показан на рис. 3.
Разность электрических потенциалов двух электродов в такой цепи называют электродвижущей силой силой цепи ЭДС(Рис. 4. Электрохимическая цепь со стандартным водородным электродом: -стандартный водородный электрод, 2-исследуемый электрод, 3 - электролитический ключ). Значение ЭДС может быть измерено, что позволяет сравнивать потенциалы электродов друг с другом. Обычно в качестве электрода, относительно которого определяют потенциалы всех систем, используют стандартный водородный электрод. Его потенциал условно принимают равным нулю.
Таким образом., электродным потенциалом называют ЭДС электрохимической цепи-гальванического элемента, составленного из исследуемого электрода и стандартного водородного электрода. Такая цепь изображена на рис. 4. Электродный потенциал обычно обозначают буквой Е.
Электрод, относительно которого производится измерение потенциала, называется электродом сравнения. Кроме водородного, в качестве электродов сравнения используют хлорсеребряный,каломельный и некоторые другие. Во всех случаях потенциал электрода сравнения принимается равным нулю. Можно перейти от одной шкалы потенциалов к другой. Например стандартный потенциал цинкового электрода по водородной шкале равен — 0,76 В, а потенциал хлорсеребряного электрода + 0,22 В (по той же шкале). Следовательно, потенциал цинкового электрода по шкале хлорсеребряного электрода будет равен: — 0,76 — 0,22 = 0,98 В. Измерение электродных потенциалов.
Точно измерить электродный потенциал достаточно трудно, так как необходимо, чтобы в процессе измерения не нарушалось равновесие на электродах. По этой причине невозможно получить точное значение Е с помощью обычного вольтметра: если мы замкнем цепь, используя вместо проводника вольтметр, то в ней начнет протекать довольно большой ток, который нарушит равновесие на электродах. Для измерения можно использовать специальные вольтметры с высоким входным сопротивлением (более 1012 Ом). При включении в цепь такого прибора протекающий ток слишком мал для оказания существенного влияния на электродное равновесие.
Стандартный электродный потенциал-это потенциал электрода при стандартных условиях, его обозначают символом Е°. Эти потенциалы определены для многих окислительно-восстановительных систем и обычно приводятся в химических справочниках. Если электроды (на пример, металлические электроды 1-го рода) расположить в порядке возрастания потенциала, то мы получим таблицу, называемую рядом стандартных электродных потенциалов. Этот ряд часто называют рядом напряжений, однако этот термин устарел и его лучше не использовать.

При помощи ряда стандартных электродных потенциалов можно характеризовать некоторые химические свойства металлов. Например, его применяют для выяснения, в какой последовательности восстанавливаются ионы металлов при электролизе, а также при описании других свойств металлов.
Чем меньше алгебраическая величина потенциала, тем выше восстановительная способностьэтого металла и тем ниже окислительная способность его ионов. Как следует из этого ряда, металлический литий - самый сильный восстановитель, а золото-самый слабый. И наоборот, ион золота Аu3+-самый сильный окислитель, а ион лития Li+ -самый слабый.
Каждый металл в ряду стандартных электродных потенциалов обладает способностью вытеснять все следующие за ним металлы из растворов их солей. Однако это не означает, что вытеснение обязательно происходит во всех случаях. Например, алюминий вытесняет медь из раствора хлорида меди (II) СuСl2, но практически не вытесняет ее из раствора сульфата меди (II) CuS04. Это объясняется тем, что хлорид-ион Сl- быстро разрушает защитную поверхностную пленку на алюминии, а сульфат-ион SO4 2-практически не разрушает ее.
Все металлы, имеющие отрицательные значения стандартных электродных потенциалов, т.е. стоящие в ряду до водорода, вытесняют водород из разбавленных кислот, анионы которых не проявляют окислительных свойств (например, из НСl или разбавленной H2S04) и растворяются в них. Однако есть и исключения. Например, свинец практически не растворяется в серной кислоте. Это обусловлено образованием на поверхности металла защитной пленки труднорастворимого сульфата свинца PbS04, который затрудняет контакт металла с раствором кислоты. Поэтому можно сделать вывод, что пользоваться рядом стандартных электродных потенциалов следует с учетом всех особенностей рассматриваемых процессов.
Стандартные потенциалы окислительно-восстановительных реакций. Возможность протекания любой окислительно-восстановительной реакции в реальных условиях обусловлена рядом причин: температурой, природой окислителя и восстановителя, кислотностью среды, концентрацией веществ, участвующих в реакции, и т. д. Учесть все эти факторы бывает трудно, но, помня о том, что любая окислительно-восстановительная реакция протекает с переносом электронов от восстановителя к окислителю, можно установить критерий возможности протекания такой реакции.
Количественной характеристикой окислительно-восстановительных процессов являются нормальные окислительно-восстановительные потенциалы окислителей и восстановителей (или стандартные потенциалы электродов).
Чтобы понять физико-химический смысл таких потенциалов, необходимо проанализировать так называемые электрохимические процессы.
Химические процессы, сопровождающиеся возникновением электрического тока или вызываемые им, называются электрохимическими.
Чтобы понять природу электрохимических процессов, обратимся к рассмотрению нескольких достаточно простых ситуаций. Представим себе металлическую пластинку, погруженную в воду. Под действием полярных молекул воды ионы металла отрываются от поверхности пластинки и гидратированными переходят в жидкую фазу. Последняя при этом заряжается положительно, а на металлической пластинке появляется избыток электронов. Чем дальше протекает процесс, тем больше становится заряд, как пластинки, так и жидкой фазы.
Благодаря электростатическому притяжению катионов раствора и избыточных электронов металла на границе раздела фаз возникает так называемый двойной электрический слой, который тормозит дальнейший переход ионов металла в жидкую фазу. Наконец, наступает момент, когда между раствором и металлической пластинкой устанавливается равновесие, которое можно выразить уравнением:
![]()
или с учетом гидратации ионов в растворе:
![]()
Состояние этого равновесия зависит от природы металла, концентрации его ионов в растворе, от температуры и давления.
При погружении металла не в воду, а в раствор соли этого металла равновесие в соответствии с принципом Ле Шателье смещается влево и тем больше, чем выше концентрация ионов металла в растворе. Активные металлы, ионы которых обладают хорошей способностью переходить в раствор, будут в этом случае заряжаться отрицательно, хотя в меньшей степени, чем в чистой воде.
Равновесие можно сместить вправо, если тем или иным способом удалять электроны из металла. Это приведет к растворению металлической пластинки. Наоборот, если к металлической пластинке подводить электроны извне, то на ней будет происходить осаждение ионов из раствора.
При погружении металла в раствор на границе раздела фаз образуется двойной электрический слой. Разность потенциалов, возникающую между металлом и окружающей его жидкой фазой, называют электродным потенциалом. Этот потенциал является характеристикой окислительно-восстановительной способности металла в виде твердой фазы.
У изолированного металлического атома (состояние одноатомного пара, возникающее при высоких температурах и высоких степенях разрежения) окислительно-восстановительные свойства характеризуются другой величиной, называемой ионизационным потенциалом. Ионизационный потенциал — это энергия, необходимая для отрыва электрона от изолированного атома.
Абсолютное значение электродного потенциала нельзя измерить непосредственно. Вместе с тем не представляет труда измерение разности электродных потенциалов, которая возникает в системе, состоящей из двух пар металл - раствор. Такие пары называют полуэлементами. Условились определять электродные потенциалы металлов по отношению к так называемому стандартному водородному электроду, потенциал которого произвольно принят за ноль. Стандартный водородный электрод состоит из специально приготовленной платиновой пластинки, погруженной в раствор кислоты с концентрацией ионов водорода 1 моль/л и омываемой струёй газообразного водорода под давлением 105 Па, при температуре 25 °С.
Ряд стандартных электродных потенциалов. Если пластинку металла, погруженную в раствор его соли с концентрацией ионов металла, равной 1 моль/л, соединить со стандартным водородным электродом, то получится гальванический элемент. Электродвижущая сила этого элемента (ЭДС), измеренная при 25 °С, и характеризует стандартный электродный потенциал металла, обозначаемый обычно как Е°.
Стандартные потенциалы электродов, выступающих как восстановители по отношению к водороду, имеют знак “-”, а знак “+” имеют стандартные потенциалы электродов, являющихся окислителями.
Металлы, расположенные в порядке возрастания их стандартных электродных потенциалов, образуют так называемый электрохимический ряд напряжений металлов: Li, Rb, К, Ва, Sr, Ca, Na, Mg, Al, Mn, Zn, Cr, Fe, Cd, Co, Ni, Sn, Pb, H, Sb, Bi, Cu, Hg, Ag, Pd, Pt, Au.
Ряд напряжений характеризует химические свойства металлов:
1. Чем более отрицателен электродный потенциал металла, тем больше его восстановительная способность.
2. Каждый металл способен вытеснять (восстанавливать) из растворов солей те металлы, которые стоят в электрохимическом ряду напряжений металлов после него.
3. Все металлы, имеющие отрицательный стандартный электродный потенциал, т. е. находящиеся в электрохимическом ряду напряжений металлов левее водорода, способны вытеснять его из растворов кислот.
Как и в случае определения значения Е° металлов, значения Е° неметаллов измеряются при температуре 25 °С и при концентрации всех атомных и молекулярных частиц, участвующих в равновесии, равной 1 моль/л.
Алгебраическое значение стандартного окислительно-восстановительного потенциала характеризует окислительную активность соответствующей окисленной формы. Поэтому сопоставление значений
стандартных окислительно-восстановительных потенциалов позволяет ответить на вопрос: протекает ли та или иная окислительно-восстановительная реакция?
Количественным критерием оценки возможности протекания той или иной окислительно-восстановительной реакции является положительное значение разности стандартных окислительно-восстановительных потенциалов полуреакций окисления и восстановления.
ВОДОРОДНЫЙ электрод в электрохимии - обычно платинированная пластина, погруженная в раствор кислоты с определенной концентрацией ионов Н+ и омываемая газообразным водородом. При давлении водорода 0,1 МПa и термодинамической активности его ионов, равной единице, потенциал водородного электрода условно принят равным нулю. Такой водородный электрод называется стандартным, он служит электродом сравнения, от которого отсчитывают потенциалы других электродов.
32 Термодинамика протекания электродных процессов. Самопроизвольность протекания окислительно-восстановительных реакций. Связь ЭДС гальванического элемента с энергией Гиббса. Связь ЭДС с константой равновесия
Любые химические реакции связаны с перемещением электронов, поэтому могут быть использованы для получения электрического тока. При этом источником электрической энергии является энергия, освобождающаяся при химической реакции. Такое превращение энергии химической реакции в электрическую возможно лишь при помощи специального устройства, называемого гальваническим элементом. Оно позволяет направлять поток электронов по металлическим проводникам.
Простое сжигание водорода сопровождается большим выделением тепла. Если его провести при постоянном объеме, например, в калориметрической бомбе, то ДU = -284,5 кДж/моль. Если эту же реакцию осуществить в гальваническом элементе электрохимическим путем, то часть этой убыли внутренней энергии может быть использована для получения электрического тока. Схема такого гальванического элемента показана на рис: IX.1. В водный раствор (например, NaOH) погружены два платиновых электрода. Левый электрод омывается пузырьками водорода, а правый - кислородом. Водород в левой части этого гальванического элемента растворяется в платине и ионизируется. Вследствие большого сродства к молекулам воды некоторое количество протонов переходит в слой раствора, непосредственно прилегающий к электроду. При этом образуются ионы гидроксония Н3О+ - они обозначены плюсами в правой части рис. IX. 1, а электроны (минусы) остаются на поверхности платинового электрода. Из-за электростатического притяжения между электронами и ионами гидроксония последние остаются вблизи электрода и не уходят в объем раствора. Благодаря этому на границе металл-раствор возникает так называемый двойной электрический слой, подобный двум обкладкам конденсатора. На поверхности правого электрода происходит реакция образования ионов гидроксила:
3/2O2г + H2Oж + 2e = 2OH-
в результате которой из металла удаляются два электрона. Поверхность металла поэтому заряжается положительно и на ней также образуется двойной электрический слой, но противоположного знака. Если соединить левый и правый электроды металлическим проводником, то по нему потечет электрический ток. Стрелка на рис. IX.1 указывает направление потока электронов. Разность электрических потенциалов на электродах разомкнутого гальванического элемента называется его электродвижущей силой (э. д. с.).
Очевидно поток электронов, возникающий в элементе может быть использован для производства работы, например, для вращения электрического мотора. Протекание тока приводит к уменьшению зарядов двойных электрических слоев. Поэтому ионы Н3О+ и ОН- получают возможность удаляться от электродов и образовывать в растворе нейтральные молекулы воды. Одновременно вследствие реакций на электродах вновь восстанавливаются двойные слои. Происходящие на электродах и в растворе изменения отражаются следующими уравнениями:
H2г = 2H+ + 2e;
3/2 O2г + H2Oж + 2e = 2OH-;
2H+ + 2OH- = 2H2Oж,
сумма которых представляет собой реакцию образования воды:
H2г + 1/2O2г = H2Oж,
Таким образом, одну и ту же реакцию образования воды из элементов можно осуществить двумя различными способами. Какой из этих способов выгоднее с точки зрения превращения энергии химической реакции в работу? В первом способе при сжигании водорода в калориметрической бомбе (V = const) при 298 К уменьшение внутренней энергии равно количеству выделившегося тепла -ДU = 284,5 кДж/моль, а работа равна нулю.
Во втором случае часть этого изменения внутренней энергии (ДG) может быть превращена в электрическую работу. Если реакция в гальваническом элементе проводится обратимо, то сопровождающая ее убыль энергии Гиббса полностью идет на производство электрической работы.
В рассматриваемом случае ДG0 = -237,2 кДж/моль и, следовательно, только ∼47 кДж/моль переходит в тепло. Этот пример показывает, что вообще энергию, освобождающуюся при горении природных видов топлива, выгоднее непосредственно преобразовывать в электрическую, так как к. п. д. тепловых машин и тепловых электростанций невелик. Описанный водородно-кислородный элемент является примером так называемых топливных элементов.
Работы по созданию таких элементов получили в последнее время широкое развитие в связи с новыми задачами техники. В этих элементах топливо и окислитель должны храниться отдельно и подаваться к электродам, на которых осуществляются электрохимические реакции. При этом элемент может работать непрерывно, если к нему подводятся реагенты и отводятся продукты реакции, что особенно удобно при использовании жидких и газообразных веществ. Вместо сжигания угля возможно использовать реакцию Ст + О2г = СО2г для получения электрического тока.
Очевидно, что в реальных условиях гальванические элементы работают необратимо, поэтому в работу превращается лишь часть изменения энергии Гиббса реакции, протекающей в элементе. Повторим, что гальванический элемент может работать при условии протекания в нем самопроизвольной химической реакции или какого-либо другого самопроизвольного процесса, сопровождающегося убылью энергии Гиббса.
Если к рассматриваемому гальваническому элементу приложить извне достаточно большую разность потенциалов, превышающую его э. д. с. и имеющую противоположное направление, то будет происходить разложение воды с выделением водорода и кислорода. Таким образом, процессы получения электрического тока в гальванических элементах и электролиза взаимно противоположны.
Особенностью электрохимического процесса в гальваническом элементе является важная для теории возможность его осуществления в условиях весьма близких к обратимости. Это достигается благодаря потенциометрическому методу, в котором э. д. с. изучаемого гальванического элемента практически полностью компенсируется с помощью противоположно направленной э.д. с. внешнего источника. Такой прием позволяет измерять э.д.с. при отсутствии тока в цепи, т.е. когда элемент не работает, а его э.д.с. максимальна. Контроль за отсутствием тока проводят гальванометрами (нуль-инструментами) высокой чувствительности. Они дают отклонение при прохождении тока силой 10-8 - 10-9 А. Такой слабый ток при прохождении через электролит даже в течение многих лет не смог бы выделить сколько-нибудь заметных количеств вещества.
Рис. IX.2. Схема измерения э.д.с. методом компенсации.
Принципиальная схема измерения э. д. с. гальванического элемента компенсационным методом показана на рис. IX.2. Постоянный ток от вспомогательной батареи ВБ подается на концы реохорда АВ - проволоки с постоянным сечением. Поэтому падение напряжения вдоль реохорда пропорционально длине соответствующего отрезка на прямой АВ. С помощью подвижного контакта С можно отбирать произвольную часть падения напряжения между точками А и В. Из рис. IX.2 видно, что напряжение, снимаемое с любого участка реохорда, например АС, направлено навстречу э. д. с. элемента X.
Передвигая контакт С по реохорду, находят такое положение, при котором нуль-гальванометр Г указывает отсутствие тока в цепи АХГС. Это означает, что падение потенциала от ВБ на отрезке АС полностью компенсирует э. д. с. элемента X.
Если э. д. с. вспомогательной батареи ВБ равна ЕБ, то э. д. с. элемента X ЕX определяется из пропорции:
ЕХ/ЕБ = АС/АВ, откуда ЕX = (АС/АВ) ЕБ.
Для того, чтобы откалибровать вспомогательную батарею перед измерениями ЕX, вместо элемента X включают другой, э. д. с. которого точно известна, например стандартный элемент Вестона. Устройство этого элемента будет описано ниже.
Повторим, что определяемая таким образом э. д. с. максимальна, так как при измерении отсутствует падение потенциала как вне, так и внутри элемента. Работа, совершаемая элементом с ничтожно малым током при обратимом проведении процесса была бы максимальной.
Теоретический и практический интерес представляют гальванические элементы с металлическими электродами. Рассмотрим, например, реакцию Znт + CuSО4водн. р-р. = ZnSО4водн. р-р + Cuт или Znт + Cu2+ = Zn+2 + +Cuт, которая может быть осуществлена двумя путями. Один из них является полностью необратимым. Цинковую пластинку помещают в водный раствор медного купороса, при этом происходит выделение металлической меди и растворение цинка. Электроны переходят от цинка непосредственно к меди, и реакция протекает без производства работы, а сопровождается только выделением тепла. В случае водородно-кислородного элемента, можно создать условия, в которых электроны будут двигаться по металлическому проводнику и совершать работу. Это достигается в гальваническом элементе, где цинковый электрод погружен в раствор ZnSO4, а медный электрод в раствор СиSO4.
Растворы отделены друг от друга пористой (керамической) перегородкой, препятствующей их смешению, но обеспечивающей прохождение электрического тока вследствие диффузии ионов через поры. Такой элемент, на электродах которого образуются двойные электрические слои, был сконструирован русским электрохимиком Б.С. Якоби.
Величина и знак электрических зарядов в двойных слоях пределяются работой удаления электрона из металла и энергией гидратации его ионов. В раствор легко будут переходить те металлы, у которых меньше работа выхода электронов и больше энергия гидратации ионов, т.е. менее благородные металлы. Так как цинк менее благороден, чем медь, то он зарядится более отрицательно по сравнению с медью. Если соединить оба электрода металлическим проводником, то электроны будут перемещаться от цинка к меди. Вследствие этого ионы цинка Zn2+ не удерживаются в двойном слое притяжением электронов, переходят в объем раствора, а перешедшие на медный электрод электроны разряжают ионы Cu2+, переводя их в металлическое состояние.
Следовательно, в процессе работы элемента происходит растворение цинкового электрода и осаждение меди на медном электроде. Чтобы элемент работал, цепь должна быть замкнутой, т.е. между растворами должен быть электрический контакт. Перенос тока внутри элемента осуществляется ионами. В элементе переход электронов от цинка к меди происходит не в условиях непосредственного контакта этих металлов, а при помощи проводника. Суммарная реакция в элементе складывается из двух пространственно разделенных электродных процессов.
Реакции, протекающие в гальванических элементах являются окислительно-восстановительными. В рассматриваемом случае окисляется цинк, который теряет электроны, а восстанавливается медь, приобретающая электроны. Вообще любая окислительно-восстановительная реакция может быть использована для получения электрического тока с помощью гальванического элемента. Как упоминалось, такой реакцией может быть горение любого вида топлива.
При схематической записи гальванических элементов границы между фазами отмечаются вертикальными линиями. При условии, что на границе двух жидкостей (в данном случае растворов ZnSO4 и CuSO4) нет разности потенциалов, ее обозначают двумя вертикальными линиями. Схема рассмотренного элемента имеет следующий вид:
Zn ∣ ZnSO4 ∥ CuSO4 ∣ Cu.
Принято записывать подобные схемы таким образом, чтобы левый электрод был отрицательным (электроны текут по металлическому проводнику слева направо и в том же направлении переносится ионами положительное электричество внутри элемента). Такая запись отвечает протеканию реакции, сопровождающейся убылью энергии Гиббса и положительной величине э. д. с.
Гальванические элементы могут быть построены не только с использованием водных растворов электролитов, но и с, применением расплавов. Примером такого элемента может служить цепь Ag ∣ AgBr ∣ Br2, в которой левый электрод серебряный, а правый - представляет собой графит, омываемый газообразным бромом, а электролитом является расплавленное AgBr. На левом электроде растворяется серебро: Agт → Ag+ + e, а на правом - адсорбированный графитом бром: 1/2Br2г + e = Br-. Таким образом, в элементе происходит реакция: Agт + 1/2Br2г = AgBrж.
В последнее время приобрели большое значение гальванические элементы с твердыми электролитами, имеющими кислородную проводимость (см. гл. VIII), например,
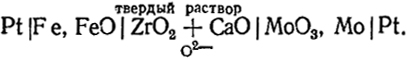
Левый электрод представляет собой смесь железа и его оксида. Здесь происходит реакция окисления железа ионами О2-, приходящими через твердый электролит. При этом освобождаются электроны, и электрод получает отрицательный заряд. На правом электроде, состоящем из смеси Мо и МоО3, происходит восстановление оксида. Это сопровождается поглощением электронов таким образом, что электрод заряжается положительно, а освободившиеся ионы О2 могут мигрировать через электролит к левому электроду. Реакция на электроде изображается следующим уравнением 3Feт + 3О2- = 3FеОт + 6е; на правом электроде: МоО3т + 6е = Мот + 3О2-.
Заметим, что сумма этих двух реакций 3Fет + МоОт = 3FеОт + Мот есть процесс восстановления оксида молибдена железом, самопроизвольное протекание которого является источником электрической энергии производимой элементом.
Из рассмотренных примеров видно, что реакцию, протекающую в гальваническом элементе, можно представить в виде двух отдельных электродных реакций.
Можно предположить, что э. д. с. гальванического элемента должна зависеть от природы реагирующих веществ, их концентраций и температуры. Чтобы найти выражения для этих зависимостей, необходимо рассмотреть термодинамические соотношения, характеризующие работу гальванического элемента.
Пусть в гальваническом элементе протекает реакция: M + Nn+ = Mn+. Работа, производимая элементом при расходе 1 моля М, определяется произведением количества электричества nF на величину э. д. с. Е, т.е. W = nFE, где п - число молей электронов, протекающих через цепь; F - число Фарадея, равное 96493 Кл. Например, для реакции Zn + Cu2+ = Zn2+ + Cu, n = 2. Если элемент работает обратимо при постоянных давлении и температуре, то произведенная им работа равна убыли энергии Гиббса, т.е. ДG = W:
ДG = -nFE = -96493E. (IX.1)
Если элемент работает необратимо, то nFE < -ДG, т.е. э.д.с. меньше, чем при обратимом проведении реакции. Выражая E в В, получаем величину ДG в Дж.
Таким образом, если известно стехиометрическое уравнение протекающей в гальваническом элементе реакции и табличные данные об изменении энергии Гиббса, можно рассчитать э. д. с.
Так, для рассмотренного выше водородно-кислородного элемента, работающего за счет энергии, освобождающейся при реакции Н2г + 1/2О2г = Н2Ож, для которой ДG 0
298 = -237200 Дж, п = 2, рH2 = рO2 = 1.
E = -ДG 0
298
/n·96493 = -(-237200/2)·96493 −∼ 1,2 В.
Из уравнения IX.1 следует, что измерение э. д. с. гальванического элемента позволяет найти изменение энергии Гиббса протекающей в нем реакции. Поэтому метод э. д. с. широко используется для определения термодинамических свойств веществ.
В приведенном выше примере этот метод позволяет найти ДG реакции восстановления МоО3 железом. Зная стандартное изменение энергии Гиббса при образовании FеО(ДG 0 f FeO) по найденному значению ДG, можно найти энергию Гиббса образования МоО3 из уравнения:
ДG = 3ДG 0
f
FeO - ДG 0
f
МоО3
Зависимость э. д. с. от температуры. Поскольку энергия Гиббса есть функция температуры, то и э. д. с. гальванического элемента также должна зависеть от температуры.
Для нахождения этой зависимости воспользуемся уравнением Гиббса-Гельмгольца: ДG = ДH + T(∂ДG/∂T)p подставив в него выражение ДG через э. д. с. При этом получим -nEF = ДH - TnF(dE/dT) или
-ДH = nF[E - Т (dE/dT)], (IX.2)
или
-ДH = W - TnF(dE/dT). (IX.3)
Сначала представим себе, что гальванический элемент, помещенный в калориметр, является коротко замкнутым. В этом случае производимая им электрическая энергия полностью превратится в тепло, количество которого равно энтальпии реакции ДH, и, следовательно, работа будет равна нулю.
Пусть теперь реакция в элементе осуществляется обратимо, например, провода от электродов выведены из калориметра, подведены к мотору, и электрический ток производит работу. Тогда часть освобождающейся при реакции энергии превратиться в электрическую работу W, а другая часть Q останется в виде тепла и будет измерена в калориметре. Согласно первому закону термодинамики
-ДH = W - Q (IX.4)
Сопоставление уравнений (IX.3) и (IX.4) показывает, что
Q = TnF(dE/dT). (IX.5)
Очевидно, чем ближе протекание реакций в гальваническом элементе к условиям обратимости, тем бо́льшая часть ДG превращается в работу. Величина Q, которая характеризует связанную энергию, определяет количество тепла, неизбежно выделяющегося (или поглощающегося) в том случае, когда элемент работает обратимо. Так как (∂ДG/∂T)р = -ДS и (∂ДG/∂Т)р = -пF(dЕ/dТ), то
ДS = nF(dE/dT), (IX.6)
и, следовательно, измерения температурной зависимости э. д. с. позволяют вычислить изменение энтропии при реакции, протекающей в гальваническом элементе. Следует подчеркнуть, что гальванический элемент может работать как с выделением, так и с поглощением тепла. В последнем случае он превращает в работу тепло окружающей среды. Это не находится в противоречии со вторым законом термодинамики, так как процессы в гальванических элементах не являются непрерывными и прекращаются при израсходовании материала электродов.
Знак и величина Q определяют температурную зависимость э. д. с. Если при работе элемента выделяется тепло, т.е. Q < 0, то температурный коэффициент э. д. с. dE/dT < 0. Это наиболее часто встречающийся случай, так как большинство элементов работает с выделением тепла. Наоборот, при Q > 0 э. д. с. растет с температурой.
Для гальванических элементов, служащих в качестве эталонов, при электрических измерениях подбирают такие реакции, в которых Q весьма мало и dЕ/dТ близко к нулю. Так, зависимость э. д. с. от температуры широко используемого стандартного элемента Вестона выражается уравнением:
E = 1,0183 - 0,0000406 (t - 20) В.
Он составлен по схеме: Cd ∣ CdSO4 ∣ ∣ Hg2SO4 ∣ Hg, и в нем протекает реакция Cdт + 2Hg+ = Cd2+ + 2Hgж1.
В качестве примера применения уравнений (IX.4) и (IX.5) вычислим величину dE/dT для элемента, в котором протекает реакция Znт + 2AgCl = ZnCl2 + 2Agт
-ДH = 217760 Дж, а E = 1,015 В при 0° C. Отсюда
-Q = -ДH = 217760 - 2·96493·1,015 = 21880 Дж.
dE/dT = -218807(273·2·96493) −∼ - 4·10-4 В/К.
Примером элемента с положительным температурным коэффициентом является ячейка Hg ∣ Hg2Cl2, KCl ∣ KOH ∣ Hg2O ∣ Hg, в которой протекает реакция Hg2Cl2 + 2KOH = 2KCl + Hg2O + H2O.
Левый электрод этого элемента называемый каломельным, часто используется в электрохимических измерениях. Он состоит из жидкой ртути, находящейся в контакте с твердой каломелью Hg2Cl2 и водным раствором какого-либо сильного электролита, например KС1. Реакция, идущая в рассматриваемом элементе, является эндотермической, ДH = 13720 Дж, а W = 31570 Дж. Таким образом Q = 13720 + 31570 = 45240 Дж, т.е. элемент поглощает из окружающей среды тепло, равное 45240 Дж. Часть этого тепла, равная 31570 Дж, идет на производство работы.
Зависимость э. д. с. от концентраций электролитов, участвующих в реакции, может быть: найдена при помощи уравнения изотермы химической реакции.
Пусть в гальваническом элементе протекает реакция A + B = 2D, при этом ДG = RTlnK + RTln (c 2 D/cAcB). Подставляя вместо ДG величину - nEF и разделив обе части уравнения на -пF, получим E = RTln(K/nF) - [RT/nFln (c 2D/cAcB)]. или, обозначая величину RTlnK/nF, зависящую только от температуры, через E0, будем иметь:
E = E0 - (RT/nF[ln(c 2 D/ cAcB)]. (IX.7.)
Величина E0 называется стандартной э. д. с. элемента. Она характеризует элемент, в котором концентрации всех участвующих в реакции веществ равны единице, а изменение энергии Гиббса равно стандартному ДG0. Заменив в уравнении (IX.7) натуральный логарифм десятичным, получим для температуры 25 °C.
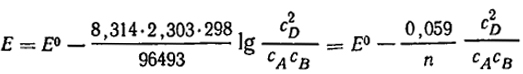
Очевидно, что для электролитов нельзя просто пользоваться аналитическими концентрациями соответствующих веществ, а необходимо учитывать диссоциацию и взаимодействие ионов. В связи с этим возникает задача определения активности электролитов.
33 Электродный потенциал. Влияние температуры и концентрации на величину электродного потенциала. Уравнение Нернста
Электродный потенциал. Уравнение Нернста
ЭДС гальванического элемента E удобно представлять в виде разности некоторых величин, характеризующих каждый из электродов – электродных потенциалов; однако для точного определения этих величин необходима точка отсчета – точно известный электродный потенциал какого-либо электрода. Электродным потенциалом электрода еэ называется ЭДС элемента, составленного из данного электрода и стандартного водородного электрода (см. ниже), электродный потенциал которого принят равным нулю. При этом знак электродного потенциала считают положительным, если в таком гальваническом элементе испытуемый электрод является катодом, и отрицательным, если испытуемый электрод является анодом. Необходимо отметить, что иногда электродный потенциал определяют как "разность потенциалов на границе электрод – раствор", т.е. считают его тождественным потенциалу ДЭС, что не вполне правильно (хотя эти величины взаимосвязаны).
Величина электродного потенциала металлического электрода зависит от температуры и активности (концентрации) иона металла в растворе, в который опущен электрод; математически эта зависимость выражается уравнением Нернста (здесь F – постоянная Фарадея, z – заряд иона):
![]() (III.40)
(III.40)
В уравнении Нернста е° – стандартный электродный потенциал, равный потенциалу электрода при активности иона металла, равной 1 моль/л. Стандартные электродные потенциалы электродов в водных растворах составляют ряд напряжений. Величина е° есть мера способности окисленной формы элемента или иона принимать электроны, т.е. восстанавливаться. Иногда различием между концентрацией и активностью иона в растворе пренебрегают, и в уравнении Нернста под знаком логарифма фигурирует концентрация ионов в растворе. Величина электродного потенциала определяет направление процесса, протекающего на электроде при работе гальванического элемента. На полуэлементе, электродный потенциал которого имеет большее (иногда говорят – более положительное) значение, будет протекать процесс восстановления, т.е. данный электрод будет являться катодом.
Рассмотрим расчёт ЭДС элемента Даниэля-Якоби с помощью уравнения Нернста. ЭДС всегда является положительной величиной и равна разности электродных потенциалов катода и анода:
![]() (III.41)
(III.41)
![]() (III.42)
(III.42)
![]() (III.43)
(III.43)
![]() (III.44)
(III.44)
![]() (III.45)
(III.45)
Как видно из уравнения (III.45), ЭДС элемента Даниэля-Якоби зависит от концентрации (точнее говоря, активности) ионов меди и цинка; при их равных концентрациях ЭДС элемента будет равна разности стандартных электродных потенциалов:
![]() (III.46)
(III.46)
Анализируя уравнение (III.45), можно определить предел необратимой работы гальванического элемента. Поскольку на аноде идет процесс окисления цинка, концентрация ионов цинка при необратимой работе гальванического элемента постоянно увеличивается; концентрация ионов меди, напротив, уменьшается. Отношение концентраций ионов меди и цинка постоянно уменьшается и логарифм этого отношения при [Сu2+] < [Zn2+] становится отрицательным. Т.о., разность потенциалов при необратимой работе гальванического элемента непрерывно уменьшается; при E = 0 (т.е. ек = еа) гальванический элемент не может совершать работу (необратимая работа гальванического элемента может прекратиться также и в результате полного растворения цинкового анода).
Уравнение (III.45) объясняет также и работоспособность т.н. концентрационных цепей – гальванических элементов, состоящих из двух одинаковых металлических электродов, опущенных в растворы соли этого металла с различными активностями а1 > а2. Катодом в этом случае будет являться электрод с большей концентрацией, т.к. стандартные электродные потенциалы обоих электродов равны; для ЭДС концентрационного гальванического элемента получаем:
![]() (III.47)
(III.47)
Единственным результатом работы концентрационного элемента является перенос ионов металла из более концентрированного раствора в менее концентрированный. Т.о., работа электрического тока в концентрационном гальваническом элементе – это работа диффузионного процесса, который проводится обратимо в результате пространственного разделения его на два противоположных по направлению обратимых электродных процесса.
В связи с тем, что потенциал электрода зависит еще от температуры и от концентрации окислителя-восстановителя в растворе, было введено понятие о стандартных электродных потенциалах (о потенциалах, измеренных при равных условиях). Это позволяет сравнивать окислительно-восстановительную способность различных веществ. Стандартные потенциалы измерены при температуре 250 С в растворах, содержащих 1 г-ион (в случае неэлектролитов по одному молю) и 1000 г. воды окисленной и восстановленной форм веществ, принимающих участие в электродной реакции. Если одна из форм вещества является газом, то раствор насыщают им под давлением 1 атм.
Так, например, стандартный редокс-потенциал электрохимической реакции Fe3+ -e < > Fe2+ равен потенциалу платиновой пластинки при 250 С в растворе, содержащем по одному г-иону ионов Fe3+ и Fe2+ в 1000 г. воды. Стандартный потенциал медного электрода равен потенциалу медной пластинки в растворе ионов Cu2+ на 1000 г. воды.
Зависимость потенциала электродной реакции
Ox + pA + ne « Red + mB
От температуры и концентрации, участвующих в ней веществ выражается формулой Нернста:
ф =ф0 + (R*T/n*F)* ln([0х]*[A]p/[Red]*[B]m)
где ф0 - стандартный электродный потенциал, в;
R – универсальная газовая постоянная Менделеева – Клапейрона, равная 8,314 Дж;
T – абсолютная температура, К;
F – число Фарадея, к;
n – число электронов, участвующих в электродной реакции;
ln – натуральный логарифм (ln х = 2,303 lg x);
[0х] и [Red] — начальные концентрации окислительной и
соответственно восстановительной формы вещества, для
которых рассчитывается окислительно-восстановительный потенциал;
[A] и [B] – начальные концентрации других веществ, участвующих
в электродной реакции (обычно вода, ионы Н+ или ОН-);
m и p – стехиометрические коэффициенты в уравнении электродной реакции.
По уравнению электродной реакции легко рассчитать окислительный потенциал системы.
34 Практическое использование электрохимических процессов. Химические источники тока
Существование электрохимических систем возможно из-за возникновения разности потенциалов между металлами и электролитом при их контакте. Измерить потенциал металла (электрода) непосредственно нельзя, но можно измерить его относительно другого электрода.
Эталоном при сопоставлении металлов по их энергетическому потенциалу является стандартный водородный электрод, потенциал которого условно принимается за нуль. Его устройство таково: платиновый электрод покрыт мелкодисперсной платиной (платиновой чернью), погружен в раствор серной кислоты с концентрацией ионов водорода 1 моль/л, обдувается струей газообразного водорода под давлением 100 кПа (Т = 298 K). Водород адсорбируется на поверхности платины. На практике при потенциометрических измерениях водородный электрод используют редко. Чаще применяют более удобные компактные электроды сравнения, имеющие определенное значение потенциала относительно водородного электрода. Обычно пользуются каломельным электродом, состоящим из металлической ртути и раствора хлорида ртути (каломели Hg2Cl2) в хлориде калия. Потенциал каломельного электрода зависит от концентрации ионов ртути, а последняя – от концентрации раствора KCl.
На основании теоретических расчетов установлено, что величина электродного потенциала, возникающая на границе между металлом и раствором соли этого металла (т. е. раствором, содержащим ионы этого металла), равна:
![]()
где Е0 – электрическая постоянная, зависящая от выбора электрода сравнения, R – газовая постоянная, равная 8,32 Дж/граджмоль, Т – абсолютная температура, n – степень окисления металла в данном соединении (в соответствии с теорией строения атома – число электронов, которое теряет атом металла, превращаясь в ион), F – число Фарадея, с – молярная концентрация ионов металла в данном растворе.
Это уравнение выражает зависимость потенциала металла от концентрации его ионов в растворе и называется уравнением Нернста. При использовании концентрированных растворов сильных электролитов концентрация иона в растворе заменяется его активностью. При активности, равной единице, второе слагаемое правой части уравнения становится равным нулю, и тогда E = E0. Если электродом сравнения взят стандартный водородный электрод, то такой гальванический элемент дает возможность получить значение стандартного электродного потенциала для данного металла.
Электродный потенциал измеряется в вольтах и равен энергии (измеряемой в джоулях, Дж), отнесенной к количеству электричества (измеряемому в кулонах, Кл), т.е. 1 В = 1 Дж/Кл. Тогда потенциалу гальванического элемента можно придать следующий физический смысл: это мера энергии, вырабатываемой в ходе протекающих в системе химических реакций. В физике единица измерения электродвижущей силы (ЭДС) – вольт – представляет собой ту силу, которая позволяет заряду в 1 кулон совершить работу в 1 джоуль.
Целостное представление о химической системе невозможно создать без связи с жизнью, с практикой. Изучение электрохимических систем необходимо для понимания не только широко используемых процессов (в гальванических элементах, в аккумуляторах, при электролизе), но и других явлений окружающего мира, в частности широко распространенных процессов коррозии. Атмосферная коррозия, разрушительное действие которой знакомо всем, возникает при контакте двух разнородных металлов, образующих гальваническую пару в среде электролита. В такой паре более активный металл играет роль анода и окисляется. Поучительной является история одного состоятельного американца, пожелавшего, не считаясь с затратами, построить уникальную яхту. Ее днище обшили дорогим монтель-металлом (сплав 70% никеля, около 30% меди; 1–2% железа и марганца), а киль, форштевень и раму руля изготовили из стали. При спуске яхты на воду в ее подводной части образовалась гальваническая пара. Значительная разность электродных потенциалов у монтель-металла и стали заставляла гальванический элемент активно работать, в результате еще до завершения отделочных работ корпус яхты дал первую течь.
Электрохимические методы широко применяются в аналитической химии. Защита окружающей среды предполагает постоянный аналитический контроль (мониторинг) множества разных объектов: во'ды (поверхностные, морские, речные, озерные), воздух (в том числе аэрозоли, пыли, туманы, дымы), почвы и донные отложения, растения, сельскохозяйственная продукция, пищевые продукты, корма, ткани животных и человека. Вредные химические вещества распространены повсюду в окружающей среде. Основная задача аналитического контроля заключается в том, чтобы получить объективную информацию о содержании вредных компонентов в среде обитания.
Начало развития электрохимических методов анализа связывают с возникновением классического электрогравиметрического метода (около 1864 г., У.Гиббс). Открытие М.Фарадеем в 1834 г. законов электролиза позднее легло в основу метода кулонометрии (применение этого метода началось с 1930-х гг.). Настоящий перелом в развитии этих методов произошел после открытия в 1922 г. Я.Гейровским метода полярографии (электролиз с капающим ртутным электродом). Электрохимические методы анализа чаще других используют в аналитической химии окружающей среды: в анализе вод, атмосферы, почв и пищи.
Таким образом, знакомясь с электрохимическими системами, учащиеся могут увидеть практическую ценность химической науки. Кроме того, рассмотрение электрохимических систем подводит учащихся к выводу о единстве важнейших явлений окружающего мира (массы и энергии, электрических явлений и химических превращений). Можно ожидать положительных результатов в развитии естественно-научного мировоззрения учащихся, если благодаря творческому подходу педагога целостность представлений об электрохимических системах будет донесена до сознания учащихся.
Химические источники тока
Химические источники тока, устройства, вырабатывающие электрическую энергию за счёт прямого преобразования химической энергии окислительно-восстановительных реакций. Первые Х. и. т. созданы в 19 в. (Вольтов столб, 1800; элемент Даниела — Якоби, 1836; Лекланше элемент, 1865, и др.). До 60-х гг. 19 в. Х. и. т. были единственными источниками электроэнергии для питания электрических приборов и для лабораторных исследований. Основу Х. и. т. составляют два электрода (один — содержащий окислитель, другой — восстановитель), контактирующие с электролитом. Между электродами устанавливается разность потенциалов — электродвижущая сила (эдс), соответствующая свободной энергии окислительно-восстановительной реакции. Действие Х. и. т. основано на протекании при замкнутой внешней цепи пространственно разделённых процессов: на отрицательном электроде восстановитель окисляется, образующиеся свободные электроны переходят по внешней цепи (создавая разрядный ток) к положительному электроду, где участвуют в реакции восстановления окислителя.
В зависимости от эксплуатационных особенностей и от электрохимической системы (совокупности реагентов и электролита) Х. и. т. делятся на гальванические элементы (обычно называются просто элементами), которые, как правило, после израсходования реагентов (после разрядки) становятся неработоспособными, и аккумуляторы, в которых реагенты регенерируются при зарядке — пропускании тока от внешнего источника (см. Зарядное устройство). Такое деление условно, т.к. некоторые элементы могут быть частично заряжены. К важным и перспективным Х. и. т. относятся топливные элементы (электрохимические генераторы), способные длительно непрерывно работать за счёт постоянного подвода к электродам новых порций реагентов и отвода продуктов реакции. Конструкция резервных химических источников тока позволяет сохранять их в неактивном состоянии 10—15 лет (см. также Источники тока).
С начала 20 в. производство Х. и. т. непрерывно расширяется в связи с развитием автомобильного транспорта, электротехники, растущим использованием радиоэлектронной и др. аппаратуры с автономным питанием. Промышленность выпускает Х. и. т., в которых преимущественно используются окислители PbO2, NiOOH, MnO2 и др., восстановителями служат Pb, Cd. Zn и др. металлы, а электролитами — водные растворы щелочей, кислот или солей (см., например, Свинцовый аккумулятор).
Основные характеристики ряда Х. и. т. приведены в табл. Лучшие характеристики имеют разрабатываемые Х. и. т. на основе более активных электрохимических систем. Так, в неводных электролитах (органических растворителях, расплавах солей или твёрдых соединениях с ионной проводимостью) в качестве восстановителей можно применять щелочные металлы (см. также Расплавные источники тока). Топливные элементы позволяют использовать энергоёмкие жидкие или газообразные реагенты.
Типы и характеристики основных первичных Химических Источников Тока
Как уже указывалось выше, к наиболее распространенным относятся марганцево-цинковые и литиевые первичные источники тока. Другие источники тока производятся в значительно меньших масштабах. Вкратце опишем основные первичные химические источники тока и их характеристики.
Марганцево-цинковые источники тока с солевым электролитом. Анодом служит цинк, являющийся корпусом источника тока, активным веществом катода - электролитический диоксид марганца или химический диоксид марганца, электролитом - хлорид аммония, хлорид цинка или хлорид аммония с хлоридом цинка. Электролит находится либо в загущенном состоянии, либо в порах микропористого сепаратора. Для снижения скорости или предотвращения коррозии в цинк и в электролит добавляют ингибиторы коррозии. К достоинствам этих батареек относятся невысокая стоимость и большое количество выпускаемых типоразмеров, к недостаткам - падающая разрядная кривая, относительно невысокая удельная энергия, значительное ухудшение характеристик при повышенных нагрузках и низких температурах. Батарейки солевые и щелочные
Марганцево-цинковые источники тока с щелочным электролитом. Анодом служит порошкообразный цинк, а катодом - диоксид марганца. Электролитом является гелеобразный раствор КОН или КОН в матрице. В состав анода и электролита включают ингибиторы коррозии. В сравнении с марганцево-цинковым источником тока с солевым электролитом батарейки с щелочным электролитом имеют более высокие емкость и удельную энергию, в особенности при повышенных нагрузках и низкой температуре, но они более дорогие. Батарейки солевые и щелочные
Ртутно-цинковые источники тока. Анодом является порошкообразный цинк, катодом - оксид ртути, электролитом - раствор КОН. Характеризуется горизонтальной разрядной кривой, высокой удельной энергией, низким саморазрядом. К недостаткам относятся плохие характеристики при пониженных температурах, высокая стоимость и, самое главное, высокая токсичность ртути. Применялись в медицинских устройствах, точных приборах и других устройствах. В последние годы из-за токсичности ртути в некоторых странах выпуск прекращен, в других странах существенно сокращен.
Ртутно-кадмиевые источники тока. Анодом служит порошкообразный кадмий, катодом - оксид ртути, электролитом - раствор КОН. Рабочие температуры окружающей среды от -55 до 80 °С. Они имеет горизонтальную разрядную кривую, очень низкий саморазряд, что обеспечивает сохранность заряда до 10 лет. Даже при температуре 60 °С саморазряд не превышает 1% в месяц. К недостаткам относятся токсичность и высокая цена компонентов. Изготавливаются в ограниченных масштабах в дисковой, цилиндрической и призматической формах. Применяются в устройствах контроля бурения нефтяных и газовых скважин, телеметрии двигателей внутреннего сгорания, сигнальных устройствах тревоги, спасательном-оборудовании, устройствах мониторинга в отдаленных районах и т.д. Из-за токсичности производство этих источников тока сокращается.
Серебряно-цинковые первичные источники тока. В качестве анода применяется порошкообразный цинк, катода - оксиды серебра, электролита - раствор КОН или NaОН (загущенные или матричные). Имеют гладкую разрядную кривую, высокую удельную энергию, низкий саморазряд, могут работать при больших токах, однако дороги. Производятся в дисковой форме емкостью до 200 мА·ч. Применяются в часах, фотоаппаратах, слуховых аппаратах и других устройствах.
Медно-цинковые источники тока. Производство этих химических источников тока началось еще в 1889 г. В настоящее время они выпускаются в небольших масштабах в виде элементов емкостью от 250 до 1000 А·ч. Гладкие цинковые пластины и пластины из смеси оксида меди, меди и связующего помещают в стеклянный или металлический сосуд с 20%-ным раствором NaОН. Элементы имеют напряжение 0,6-0,7 В и удельную энергию 25-30 Вт·ч/кг. К их достоинствам относится постоянство разрядного напряжения, очень малый саморазряд, безотказность в работе и невысокая цена. Применялись в системах сигнализации и связи на железных дорогах.
Воздушно-цинковые первичные источники тока. Активным веществом катода служит кислород воздуха, поэтому катод является нерасходуемым, он содержит катализатор восстановления кислорода (активированный уголь или диоксид марганца). В качестве электролита применяется раствор КОН. К достоинствам источника тока относятся очень высокая удельная энергия и относительно невысокая цена, к недостаткам - влияние окружающей среды (влажности воздуха и диоксида углерода) на характеристики источника тока. Производятся две разновидности: призматические с высокой емкостью (до 1000 А·ч) и дисковые с малой емкостью. Используются для питания средств связи, в слуховых аппаратах, медицинских и других устройствах.
Литиевые первичные источники тока с твердыми катодами и апротонным электролитом. Восстановителем является литий, окислителями - оксиды, сульфиды металлов или фтороуглерод. Электролитами служат растворы солей лития (LiClO4, LiBF4 или LiBr) в апротонных растворителях: пропиленкарбонате (ПК), диоксолане (ДОЛ), г-бутиролактоне (БЛ), тетрагидрофуране (ТГФ), диметоксиэтане (ДМЭ) и др. В зависимости от типа используемого окислителя источник тока имеет разрядное напряжение около 1,5В (CuO, CuS, FeS, Bi2O3 или FeS2) или 2,5-3,2В (MnO2, (CF)n, Ag2V4O11, Ag2CrO4, Cu4O(PO4)2 и др.). Литиевые первичные источники тока имеют более высокую емкость и удельную энергию, более широкий интервал рабочих температур, лучшую работоспособность при пониженных температурах и меньшую скорость саморазряда по сравнению с этими же параметрами марганцево-цинковых источников тока. Однако они дороже марганцево-цинковых элементов. Литиевые источники тока с напряжением 1,5В заменяют марганцево-цинковые батарейки одинакового типоразмера, источники тока с напряжением 2,5-3,2В заменяют батареи марганцево-цинковых элементов. Они используются в медицинской, бытовой, промышленной и военной электронике. Литиевые Батарейки
Литиевые источники тока с жидким или растворенным окислителем. В этих источниках тока используются диоксид серы (SO2), растворяющийся в органическом растворителе, жидкие тионилхлорид (SOCl2) и сульфурилхлорид (SO2Cl2). Катоды в источнике тока нерастворимые и изготавливаются из углеродистых материалов, нанесенных на алюминиевую (для SO2), никелевую основу или нержавеющую сталь. Электролитом в элементе системы литий - диоксид серы является LiBr, растворенный в ацетонитриле, в элементах с тионилхлоридом и сульфурилхлоридом - LiAlCl4 в SOCl2 или в SO2Cl2 с добавками. Эти источники тока имеют очень высокую удельную энергию, высокие скорости разряда и удельную мощность, горизонтальную разрядную кривую, способность функционировать при низких температурах (до -55 °С), длительный ресурс. К недостаткам следует отнести сравнительно высокую стоимость, работу под давлением, потенциальную взрывоопасность, присутствие токсичных компонентов. Используются в тех областях, где требуются высокие удельная энергия и мощность, длительная сохранность, способность работать при низких температурах (в космической и военной технике, системах сохранения памяти, и других устройствах). Литиевые Батарейки
Йодно-литиевые источники тока с твердым электролитом. Окислителем является йод, растворенный в твердом поливинилпиридине (ПВП), электролитом - твердая соль LiI, толщина которой непрерывно возрастает в результате токообразующей реакции. Эти источники тока могут храниться очень продолжительное время, имеют высокую удельную энергию, широкий диапазон рабочих температур, но очень низкие скорость разряда и удельную мощность. Используются в основном в кардиостимуляторах и производятся для этих целей в специальной D-образной форме. Литиевые Батарейки
35. Коррозия металлов. Основные виды коррозии. Химическая коррозия
Термин коррозия происходит от латинского слова corrodere, что означает разъедать, разрушать.
Коррозия – это самопроизвольный процесс разрушения материалов и изделий из них под химическим воздействием окружающей среды.
Коррозия металлов – разрушение металлов вследствие физико-химического воздействия внешней среды, при котором металл переходит в окисленное (ионное) состояние и теряет присущие ему свойства.
В тех случаях, когда окисление металла необходимо для осуществления какого-либо технологического процесса, термин «коррозия» употреблять не следует. Например, нельзя говорить о коррозии растворимого анода в гальванической ванне, поскольку анод должен окислятся, посылая свои ионы в раствор, чтобы протекал нужный процесс. Нельзя также говорить о коррозии алюминия при осуществлении алюмотермического процесса. Но физико-химическая сущность изменений, происходящих с металлом во всех подобных случаях, одинакова: металл окисляется.
Классификация коррозионных сред
Среда, в которой металл подвергается коррозии (коррозирует) называется коррозионной или агрессивной средой. По степени воздействия на металлы коррозионные среды целесообразно разделить на:
неагрессивные;
слабоагрессивные;
среднеагрессивные;
сильноагрессивные.
Для определения степени агрессивности среды при атмосферной коррозии необходимо учитывать условия эксплуатации металлических конструкций зданий и сооружений. Степень агрессивности среды по отношению к конструкциям внутри отапливаемых и неотапливаемых зданий, зданий без стен и постоянно аэрируемых зданий определяется возможностью конденсации влаги, а также температурно-влажностным режимом и концентрацией газов и пыли внутри здания. Степень агрессивности среды по отношению к конструкциям на открытом воздухе, не защищенным от непосредственного попадания атмосферных осадков, определяется климатической зоной и концентрацией газов и пыли в воздухе. С учетом влияния метеорологических факторов и агрессивности газов разработана классификация степени агрессивности сред по отношению к строительным металлическим конструкциям. С учетом влияния метеорологических факторов и агрессивности газов разработана классификация степени агрессивности сред по отношению к строительным металлическим конструкциям.
Таким образом, защита металлических конструкций от коррозии определяется агрессивностью условий их эксплуатации. Наиболее надежными защитными системами металлических конструкций являются алюминиевые и цинковые покрытия.
Классификация коррозионных процессов
По типу разрушений
По типу разрушений коррозия бывает сплошной и местной.
При равномерном распределении коррозионных разрушений по всей поверхности металла коррозию называют равномерной или сплошной. Она не представляет собой опасности для конструкций и аппаратов, особенно в тех случаях, когда потери металлов не превышают технически обоснованных норм. Её последствия могут быть сравнительно легко учтены.
Если же значительная часть поверхности металла свободна от коррозии и последняя сосредоточена на отдельных участках, то ее называют местной. Она гораздо опаснее, хотя потери металла могут быть и небольшими. Её опасность состоит в том, что, снижая прочность отдельных участков, она резко уменьшает надёжность конструкций, сооружений, аппаратов. Местной коррозии благоприятствуют морская вода, растворы солей, в частности галогенидных: хлорид натрия, кальция, магния. Особенно большие неприятности связаны с хлоридом натрия, который разбрасывают в зимнее время на дорогах и тротуарах для удаления снега и льда. В присутствии солей они плавятся, и образующиеся растворы стекают в канализационные трубы. Соли являются активаторами коррозии и приводят к ускоренному разрушению металлов, в частности транспортных средств и подземных коммуникаций. Подсчитано, что в США применение для этой цели солей приводит к потерям на сумму 2 млрд. долларов в год в связи с коррозией двигателей и 0,5 млрд. на дополнительный ремонт дорог, подземных магистралей и мостов. Причина же использования хлорида натрия заключается в его дешевизне. В настоящее время выход лишь один – вовремя убирать снег и вывозить его на свалки. Экономически он белее чем оправдан.
Язвенная (в виде пятен различной величины), точечная, щелевая, контактная, межкристаллическая коррозия - наиболее часто встречающиеся в практике типы местной коррозии. Точечная - одна из наиболее опасных. Она заключается в образовании сквозных поражений, то есть точечных полостей – питтингов.
Коррозионное растрескивание возникает при одновременном воздействии на металл агрессивной среды и механических напряжений. В металле появляются трещины транскристаллитного характера, которые часто приводят к полному разрушению изделий.
По механизму
По механизму коррозионного процесса различают два основных типа коррозии: химическую и электрохимическую. Строго отделить один вид от другого трудно, а иногда и невозможно.
Под химической коррозией подразумевают взаимодействие металлической поверхности с окружающей средой, не сопровождающееся возникновением электрохимических (электродных) процессов на границе фаз. Она основана на реакции между металлом и агрессивным реагентом. Этот вид коррозии протекает в основном равномерно по всей поверхности металла. В связи с этим химическая коррозия менее опасна, чем электрохимическая.
Примером химической коррозии служат ржавление железа и покрытие патиной бронзы. В промышленном производстве металлы нередко нагреваются до высоких температур. В таких условиях химическая коррозия ускоряется. Многие знают, что на прокатке раскаленных кусков металла образуется окалина. Это типичный продукт химической коррозии.
Установлено, что коррозии железа способствует наличие в нём серы. Античные предметы, изготовленные из железа, устойчивы к коррозии именно благодаря низкому содержанию в этом железе серы. Сера в железе обычно содержится в виде сульфидов FeS и других. В процессе коррозии сульфиды разлагаются с выделением сероводорода H2S, который является катализатором коррозии железа.
Механизм химической коррозии сводится к реактивной диффузии атомов или ионов металла сквозь постепенно утолщающуюся пленку продуктов коррозии (например, окалины) и встречной диффузии атомов или ионов кислорода. По современным воззрениям этот процесс имеет ионно-электронный механизм, аналогичный процессам электропроводности в ионных кристаллах.
Особенно разнообразные процессы химической коррозии встречаются в различных производствах. В атмосфере водорода, метана и других углеводородов, оксида углерода (II), сероводорода, хлора, в среде кислот, а также в расплавах солей и других веществ протекают специфические реакции с вовлечением материала аппаратов и агрегатов, в которых осуществляется химический процесс. Задача специалистов при конструировании реактора – подобрать металл или сплав, который был бы наиболее устойчив к компонентам химического процесса.
Практически наиболее важным видом химической коррозии является взаимодействие металла при высоких температурах с кислородом и другими газообразными активными средами (HS, SO, галогены, водяные пары, CO). Подобные процессы химической коррозии металлов при повышенных температурах носят также название газовой коррозии. Многие ответственные детали инженерных конструкций сильно разрушаются от газовой коррозии (лопатки газовых турбин, сопла ракетных двигателей, элементы электронагревателей, колосники, арматура печей). Большие потери от газовой коррозии (угар металла) несет металлургическая промышленность. Стойкость против газовой коррозии повышается при введении в состав сплава различных добавок (хрома, алюминия, кремния). Добавки алюминия, бериллия и магния к меди повышают ее сопротивление газовой коррозии в окислительных средах. Для защиты железных и стальных изделий от газовой коррозии поверхность изделия покрывают алюминием (алитирование).
Под электрохимической коррозией подразумевают процесс взаимодействия металлов с электролитами в виде водных растворов, реже с неводными электролитами, например, с некоторыми органическими электропроводными соединениями или безводными расплавами солей при повышенных температурах.
Электрохимическая коррозия часто связана с наличием в металле случайных примесей или специально введенных легирующих добавок.
Многие химики в своё время были озадачены тем, что иногда реакция
Zn + H2SO4 = ZnSO4 + H2
не протекает. Было выяснено, что в такой ситуации в раствор нужно добавить немного сульфата меди (II) (медного купороса). В этом случае на поверхности цинка выделится медь
CaSO4 + Zn = ZnSO4 + Cu
и водород начнёт бурно выделяться. При объяснении данного явления в 1830 году швейцарским химиком А. де-ля Ривом была создана первая электрохимическая теория коррозии.
В 1800 году, вскоре после открытия итальянцем Л. Гальвани электрохимического явления, его соотечественник А. Вольта сконструировал источник электрического тока – гальванический элемент, что открыло человечеству эру электричества. В одном из вариантов источник состоял из чередующихся медных и цинковых дисков, разделенных пористым материалом и пропитанных раствором соли. В зависимости от числа дисков получается ток различной силы. При осаждении на поверхности цинка металлической меди получается короткозамкнутый элемент. В нём цинк является анодом, а медь – катодом. Поскольку медь находится в контакте с цинком и оба эти металла окружены раствором электролита, гальванический элемент является «включенным». Цинк в виде иона Zn2+ переходит в раствор серной кислоты, а оставшиеся от каждого атома два электрона перетекают на более электроположительный металл – медь:
Zn = Zn2+ + 2e–
К медному аноду подходят ионы водорода, принимают электроны и превращаются в атомы водорода, а затем и в молекулы водорода:
H+ + e (Cu) = H
2H = H2
Таким образом, потоки движения ионов разделены и при избытке кислоты процесс протекает до тех пор, пока не растворится весь цинк.
Итак, процессы электрохимической коррозии протекают по законам электрохимической кинетики, когда общая реакция взаимодействия может быть разделена на следующие, в значительной степени самостоятельные, электродные процессы:
анодный процесс - переход металла в раствор в виде ионов (в водных растворах, обычно гидратированных) с оставлением эквивалентного количества электронов в металле;
катодный процесс - ассимиляция появившихся в металле избыточных электронов деполяризаторами.
Различают коррозию с водородной, кислородной или окислительной деполяризацией. При наличии в растворе газообразного кислорода и невозможностью протекания процесса коррозии с водородной деполяризацией основную роль деполяризатора исполняет кислород. Коррозионные процессы, у которых катодная деполяризация осуществляется растворенным в электролите кислородом, называют процессами коррозии металлов с кислородной деполяризацией. Это наиболее распространенный тип коррозии металла в воде, в нейтральных и даже в слабокислых солевых растворах, в морской воде, в земле, в атмосфере воздуха.
Общая схема кислородной деполяризации сводится к восстановлению молекулярного кислорода до иона гидроокисла:
O + 4e +2HO 4OH
Коррозия металла с кислородной деполяризацией в большинстве практических случаев происходит в электролитах, соприкасающихся с атмосферой, парциальное давление кислорода в которой равно 0,21 атм.
Каждый процесс с кислородной деполяризацией включает следующие последовательные стадии.
Растворение кислорода в электролите.
Транспортировка растворенного кислорода в растворе электролита (за счет диффузии или перемешивания).
Перенос кислорода в результате движения электролита.
Перенос кислорода в диффузионном слое электролита или в пленке продуктов коррозии металла к катодным участкам поверхности.
Ионизация кислорода:
В реальных условиях коррозии металла наиболее затрудненными стадиями процесса являются:
Реакция ионизации кислорода на катоде. Возникающую при этом поляризацию называют перенапряжением кислорода. Говорят, что процесс идет с кинетическим контролем.
Диффузия кислорода к катоду, либо перенапряжение диффузии. В этом случае, говорят, что процесс идет с диффузионным контролем.
Возможны случаи, когда обе стадии – ионизация кислорода и диффузия кислорода оказывают влияние на процесс. Тогда говорят, о кинетически-диффузионном контроле.
Сущность первой электрохимической теории состояла в том, что примеси в металлах создают микрогальванические элементы, в которых происходит перетекание электронов от анодных участков к катодным. Поскольку катодный и анодный процессы разделены на поверхности, то разделены и противоположные потоки ионов, атомов и молекул. Разделенные потоки не мешают друг другу, и по этой причине процесс коррозии протекает быстрее, чем в случае микрогальванических элементов.
Конечно, в настоящее время теории электрохимической коррозии выглядят гораздо более совершенными. Они основаны на многочисленных экспериментальных фактах и выражены в математической форме.
Различают следующие типы электрохимической коррозии, имеющие наиболее важное практическое значение.
1. Коррозия в электролитах. К этому типу относятся коррозия в природных водах (морской и пресной), а также различные виды коррозии в жидких средах. В зависимости от характера среды различают:
а) кислотную;
б) щелочную;
в) солевую;
г) морскую коррозию.
По условиям воздействия жидкой среды на металл этот тип коррозии также характеризуется как:
коррозия при полном погружении;
при неполном погружении;
при переменном погружении.
КОРРОЗИЯ МЕТАЛЛОВ ХИМИЧЕСКАЯ
— разрушение металлов вследствие хим. взаимодействия их с агрессивной средой, не проводящей электрич. ток. Процесс химической коррозии металлов характеризуется прямым соединением металла с агрессивными составными частями среды. Напр., железо при нагревании до высоких темп-р в атмосфере воздуха или печных топочных газов окисляется кислородом в продукты, наз. окалиной. Химическая коррозия металлов в газах при высоких темп-pax, называемая газовой коррозией, — сравнительно простой вид коррозии. Скорость К.м.х. в этом случае определяется в осн. свойствами слоя продуктов коррозии (защитной пленкой), сцепл. с поверхностью металла и возникающего в результате самого корроз. процесса. Свойства возникающих на металле защитных пленок зависят от состава металла, среды и условий (темп-ры, времени, скорости движения среды и др.). Введение в сталь хрома, алюминия, кремния значительно повышает ее стойкость против газовой коррозии. Химическая коррозия теп-лоэнергетич. оборудования протекает под действием насыщ. и перегретого пара. При работе оборудования хим. равновесие в реакции между водяным паром и железом не достигается, т.к. при парообразовании непрерывно отводится водород — один из продуктов реакции. Значит, корроз. стойкость металла теплоэнергетич. оборудования объясняется наличием на поверхности железа относительно коррозионно-стойкого защитного слоя, к-рый образуется при эксплуатации и состоит из закиси-окиси железа.
36. Коррозия металлов. Электрохимическая коррозия
Корро́зия (от лат. corrosio — разъедание) — это самопроизвольное разрушение металлов в результате химического или физико-химического взаимодействия с окружающей средой. В общем случае это разрушение любого материала, будь то металл или керамика, дерево или полимер. Причиной коррозии служит термодинамическая неустойчивость конструкционных материалов к воздействию веществ, находящихся в контактирующей с ними среде. Пример — кислородная коррозия железа в воде: 4Fe + 6Н2О + ЗО2 = 4Fe(OH)3. Гидратированный оксид железа Fe(OН)3 и является тем, что называют ржавчиной.
В повседневной жизни для сплавов железа (сталей) чаще используют термин «ржавление». Менее известны случаи коррозии полимеров. Применительно к ним существует понятие «старение», аналогичное термину «коррозия» для металлов. Например, старение резины из-за взаимодействия с кислородом воздуха или разрушение некоторых пластиков под воздействием атмосферных осадков, а также биологическая коррозия. Скорость коррозии, как и всякой химической реакции, очень сильно зависит от температуры. Повышение температуры на 100 градусов может увеличить скорость коррозии на несколько.
Классификация видов коррозии
Коррозионные процессы отличаются широким распространением и разнообразием условий и сред, в которых они протекают. Поэтому пока нет единой и всеобъемлющей классификации встречающихся случаев коррозии.
По типу агрессивных сред, в которых протекает процесс разрушения, коррозия может быть следующих видов:
газовая коррозия;
атмосферная коррозия;
коррозия в неэлектролитах;
коррозия в электролитах;
подземная коррозия;
биокоррозия;
коррозия под воздействием блуждающих токов.
По условиям протекания коррозионного процесса различаются следующие виды:
контактная коррозия;
щелевая коррозия;
коррозия при неполном погружении;
коррозия при полном погружении;
коррозия при переменном погружении;
коррозия при трении;
межкристаллитная коррозия;
коррозия под напряжением.
По характеру разрушения:
сплошная коррозия, охватывающая всю поверхность:
равномерная;
неравномерная;
избирательная[1];
локальная (местная) коррозия, охватывающая отдельные участки:
пятнами;
язвенная;
точечная (или питтинг);
сквозная;
межкристаллитная (расслаивающая в деформированных заготовках и ножевая в сварных соединениях).
Главная классификация производится по механизму протекания процесса. Различают два вида:
химическую коррозию;
электрохимическую коррозию.
Коррозия металлов
Образование гальванических пар с пользой применяют для создания батарей и аккумуляторов. С другой стороны, образование такой пары приводит к неблагоприятному процессу, жертвой которого становится целый ряд металлов, — коррозии. Под коррозией понимают происходящее на поверхности электрохимическое или химическое разрушение металлического материала. Наиболее часто при коррозии металл окисляется с образованием ионов металла, которые при дальнейших превращениях дают различные продукты коррозии. Коррозия может быть вызвана как химическим, так и электрохимическим процессом. Соответственно, различают химическую и электрохимическую коррозию металлов.
Электрохимическая коррозия
Разрушение металла под воздействием возникающих в коррозионной среде гальванических элементов называют электрохимической коррозией. Не следует путать с электрохимической коррозией коррозию однородного материала, например, ржавление железа или т.п. При электрохимической коррозии (наиболее частая форма коррозии) всегда требуется наличие электролита (Конденсат, дождевая вода и т. д.), с которым соприкасаются электроды - либо различные элементы структуры материала, либо два различных соприкасающихся материала с различающимися окислительно-восстановительными потенциалами. Если в воде растворены ионы солей, кислот, или т.п., электропроводность ее повышается, и скорость процесса увеличивается
При соприкосновении двух металлов с различными окислительно-восстановительными потенциалами и погружении их в раствор электролита, например, дождевой воды с растворенным углекислым газом CO2, образуется гальванический элемент, так называемый коррозионный элемент. Он представляет собой не что иное, как замкнутую гальваническую ячейку. В ней происходит медленное растворение металлического материала с более низким окислительно-восстановительным потенциалом; второй электрод в паре, как правило, не корродирует. Этот вид коррозии особо присущ металлам с высокими отрицательными потенциалами. Так, совсем небольшого количества примеси на поверхности металла с большим редокспотенциалом уже достаточно для возникновения коррозионного элемента. Особо подвержены риску места соприкосновения металлов с различными потенциалами, например, сварочные швы или заклёпки.
Если растворяющийся электрод коррозионно-стоек, процесс коррозии замедляется. На этом основана, например, защита железных изделий от коррозии путём лужения или оцинковки - олово или цинк имеют более отрицательный потенциал, чем железо, поэтому в такой паре железо восстанавливается, а олово или цинк должны корродировать. Однако в связи с образованием на поверхности олова или цинка оксидной плёнки процесс коррозии сильно замедляется.
37. Методы защиты металлов от коррозии: изменение свойств коррозионной среды, защитные покрытия, электрохимическая защита
МЕТОДЫ ЗАЩИТЫ ОТ КОРРОЗИИ
Проблема защиты металлов от коррозии возникла почти в самом начале их использования. Люди пытались защитить металлы от атмосферного воздействия с помощью жира, масел, а позднее и покрытием другими металлами и, прежде всего, легкоплавким оловом. В трудах древнегреческого историка Геродота (V век до нашей эры) уже имеется упоминание о применении олова для защиты железа от коррозии
Задачей химиков было и остается выяснение сущности явлений коррозии, разработка мер, препятствующих или замедляющих её протекание. Коррозия металлов осуществляется в соответствии с законами природы и поэтому ее нельзя полностью устранить, а можно лишь замедлить.
В зависимости от характера коррозии и условий ее протекания применяются различные методы защиты. Выбор того или иного способа определяется его эффективностью в данном конкретном случае, а также экономической целесообразностью.
Легирование
Имеется способ уменьшения коррозии металлов, который строго нельзя отнести к защите. Этим способом является получение сплавов, которое называется легирование. В настоящее время создано большое число нержавеющих сталей путем присадок к железу никеля, хрома, кобальта и др. Такие стали, действительно, не покрываются ржавчиной, но их поверхностная коррозия имеет место, хотя и с малой скоростью. Оказалось, что при использовании легирующих добавок коррозионная стойкость меняется скачкообразно. Установлено правило, названное правилом Таммана, согласно которому резкое повышение устойчивости к коррозии железа наблюдается при введении легирующей добавки в количестве 1/8 атомной доли, то есть один атом легирующей добавки приходится на восемь атомов железа. Считается, что при таком соотношении атомов происходит их упорядоченное расположение в кристаллической решетке твердого раствора, что и затрудняет коррозию.
Защитные пленки
Одним из наиболее распространенных способов защиты металлов от коррозии является нанесение на их поверхность защитных пленок: лака, краски, эмали, других металлов. Лакокрасочные покрытия наиболее доступны для широкого круга людей. Лаки и краски обладают низкой газо- и паропроницаемостью, водоотталкивающими свойствами, поэтому они препятствуют доступу к поверхности металла воды, кислорода и содержащихся в атмосфере агрессивных компонентов. Покрытие поверхности металла лакокрасочным слоем не исключает коррозию, а служит для нее лишь преградой, а значит, лишь тормозит процесс коррозии. Именно поэтому важное значение имеет качество покрытия – толщина слоя, пористость, равномерность, проницаемость, способность набухать в воде, прочность сцепления (адгезия). Качество покрытия зависит от тщательности подготовки поверхности и способа нанесения защитного слоя. Окалина и ржавчина должны быть удалены с поверхности покрываемого металла. В противном случае они будут препятствовать хорошей адгезии покрытия с поверхностью металла. Низкое качество покрытия нередко связано с повышенной пористостью. Часто она возникает в процессе формирования защитного слоя в результате испарения растворителя и удаления продуктов отверждения и деструкции (при старении пленки). Поэтому обычно рекомендуют наносить не один толстый слой, а несколько тонких слоев покрытия. Во многих случаях увеличение толщины покрытия приводит к ослаблению адгезии защитного слоя с металлом. Большой вред наносят воздушные полости, пузыри. Они образуются при низком качестве выполнения операции нанесения покрытия.
Для снижения смачиваемости водой лакокрасочные покрытия иногда, в свою очередь, защищают восковыми составами или кремнийорганическими соединениями. Лаки и краски наиболее эффективны для защиты от атмосферной коррозии. В большинстве случаев они непригодны для защиты подземных сооружений и конструкций, так как трудно предупредить механические повреждения защитных слоев при контакте с грунтом. Опыт показывает, что срок службы лакокрасочных покрытий в этих условиях невелик. Намного практичнее оказалось применять толстослойные покрытия из каменноугольной смолы (битума).
В некоторых случаях пигменты красок выполняют также роль ингибиторов коррозии (об ингибиторах будет сказано далее). К числу таких пигментов относятся хроматы стронция, свинца и цинка (SrCrO4, PbCrO4, ZnCrO4).
Грунтовки и фосфатирование
Часто под лакокрасочный слой наносят грунтовки. Пигменты, входящие в ее состав, также должны обладать ингибиторными свойствами. Проходя через слой грунтовки, вода растворяет некоторое количество пигмента и становится менее коррозионноактивной. Среди пигментов, рекомендуемых для грунтов, наиболее эффективным признан свинцовый сурик Pb3O4.
Вместо грунтовки иногда проводят фосфатирование поверхности металла. Для этого на чистую поверхность кистью или распылителем наносят растворы ортофосфатов железа (III), марганца (II) или цинка (II), содержащих и саму ортофосфорную кислоту H3PO4. В заводских условиях фосфатирование ведут при 99-970 С в течение 30-90 минут. В образование фосфатного покрытия вносят вклад металл, растворяющийся в фосфатирующейся смеси, и оставшиеся на его поверхности оксиды.
Для фосфатирования поверхности стальных изделий разработано несколько различных препаратов. Большинство из них состоят из смеси фосфатов марганца и железа. Возможно, наиболее распространенным препаратом является «мажеф» – смесь дигидрофосфатов марганца Mn(H2PO4)2, железа Fe(H2PO4)2 и свободной фосфорной кислоты. Название препарата состоит из первых букв компонентов смеси. По внешнему виду мажеф – это мелкокристаллический порошок белого цвета с соотношением между марганцем и железом от 10:1 до 15:1. Он состоит из 46-52% P2O5; не менее 14% Mn; 0,3-3% Fe. При фосфатировании мажефом стальное изделие помещается в его раствор, нагретый примерно до ста градусов. В растворе происходит растворение с поверхности железа с выделением водорода, а на поверхности образуется плотный, прочный и малорастворимый в воде защитный слой фосфатов марганца и железа серо-черного цвета. При достижении толщины слоя определенной величины дальнейшее растворение железа прекращается. Пленка фосфатов защищает поверхность изделия от атмосферных осадков, но мало эффективна от растворов солей и даже слабых растворов кислот. Таким образом, фосфатная пленка может служить лишь грунтом для последовательного нанесения органических защитных и декоративных покрытий – лаков, красок, смол. Процесс фосфатирования длится 40-60 минут. Для его ускорения в раствор вводят 50-70 г/л нитрата цинка. В этом случае время сокращается в 10-12 раз.
Электрохимическая защита
В производственных условиях используют также электрохимический способ – обработку изделий переменным током в растворе фосфата цинка при плотности тока 4 А/дм2 и напряжении 20 В и при температуре 60-700 С. Фосфатные покрытия представляют собой сетку плотносцепленных с поверхностью фосфатов металлов. Сами по себе фосфатные покрытия не обеспечивают надежной коррозионной защиты. Преимущественно их используют как основу под окраску, обеспечивающую хорошее сцепление краски с металлом. Кроме того, фосфатный слой уменьшает коррозионные разрушения при образовании царапин или других дефектов.
Силикатные покрытия
Для защиты металлов от коррозии используют стекловидные и фарфоровые эмали, коэффициент теплового расширения которых должен быть близок к таковому для покрываемых металлов. Эмалирование осуществляют нанесением на поверхность изделий водной суспензии или сухим напудриванием. Вначале на очищенную поверхность наносят грунтовочный слой и обжигают его в печи. Далее наносят слой покровной эмали и обжиг повторяют. Наиболее распространены стекловидные эмали – прозрачные или загашенные. Их компонентами являются SiO2 (основная масса), B2O3, Na2O, PbO. Кроме того, вводят вспомогательные материалы: окислители органических примесей, оксиды, способствующие сцеплению эмали с эмалируемой поверхностью, глушители, красители. Эмалирующий материал получают сплавлением исходных компонентов, измельчением в порошок и добавлением 6-10% глины. Эмалевые покрытия в основном наносят на сталь, а также на чугун, медь, латунь и алюминий.
Эмали обладают высокими защитными свойствами, которые обусловлены их непроницаемостью для воды и воздуха (газов) даже при длительном контакте. Их важным качеством является высокая стойкость при повышенных температурах. К основным недостаткам эмалевых покрытий относят чувствительность к механическим и термическим ударам. При длительной эксплуатации на поверхности эмалевых покрытий может появиться сетка трещин, которая обеспечивает доступ влаги и воздуха к металлу, вследствие чего и начинается коррозия.
Цементные покрытия
Для защиты чугунных и стальных водяных труб от коррозии используют цементные покрытия. Поскольку коэффициенты теплового расширения портландцемента и стали близки, то он довольно широко применяется для этих целей. Недостаток портландцементных покрытий тот же, что и эмалевых, – высокая чувствительность к механическим ударам.
Покрытие металлами
Широко распространенным способом защиты металлов от коррозии является покрытие их слоем других металлов. Покрывающие металлы сами корродируют с малой скоростью, так как покрываются плотной оксидной пленкой. Покрывающий слой наносят различными методами:
горячее покрытие – кратковременное погружение в ванну с расплавленным металлом;
гальваническое покрытие – электроосаждение из водных растворов электролитов;
металлизация – напыление;
диффузионное покрытие – обработка порошками при повышенной температуре в специальном барабане;
с помощью газофазной реакции, например:
3CrCl2 + 2Fe 1000 ‘ C 2FeCl3 + 3Cr (в расплаве с железом).
Имеются и другие методы нанесения металлических покрытий. Например, разновидностью диффузионного способа является погружение изделий в расплав хлорида кальция, в котором растворены наносимые металлы.
В производстве широко используется химическое нанесение металлических покрытий на изделия. Процесс химического металлирования является каталитическим или автокаталитическим, а катализатором является поверхность изделия. Используемый раствор содержит соединение наносимого металла и восстановитель. Поскольку катализатором является поверхность изделия, выделение металла и происходит именно на ней, а не в объеме раствора. В настоящее время разработаны методы химического покрытия металлических изделий никелем, кобальтом, железом, палладием, платиной, медью, золотом, серебром, родием, рутением и некоторыми сплавами на основе этих металлов. В качестве восстановителей используют гипофосфит и боргидрид натрия, формальдегид, гидразин. Естественно, что химическим никелированием можно наносить защитное покрытие не на любой металл.
Металлические покрытия делят на две группы:
коррозионностойкие;
протекторные.
Например, для покрытия сплавов на основе железа в первую группу входят никель, серебро, медь, свинец, хром. Они более электроположительны по отношению к железу, то есть в электрохимическом ряду напряжений металлов стоят правее железа. Во вторую группу входят цинк, кадмий, алюминий. Они более электроотрицательны по отношению к железу.
В повседневной жизни человек чаще всего встречается с покрытиями железа цинком и оловом. Листовое железо, покрытое цинком, называют оцинкованным железом, а покрытое оловом – белой жестью. Первое в больших количествах идет на кровли домов, а второе – на изготовление консервных банок. Впервые способ хранения пищевых продуктов в жестяных банках предложил повар Н.Ф. Аппер в 1810 году. И то, и другое железо получают, главным образом, протягиванием листа железа через расплав соответствующего металла.
Металлические покрытия защищают железо от коррозии при сохранении сплошности. При нарушении же покрывающего слоя коррозия изделия протекает даже более интенсивно, чем без покрытия. Это объясняется работой гальванического элемента железо–металл. Трещины и царапины заполняются влагой, в результате чего образуются растворы, ионные процессы в которых облегчают протекание электрохимического процесса (коррозии).
Ингибиторы
Применение ингибиторов – один из самых эффективных способов борьбы с коррозией металлов в различных агрессивных средах. Ингибиторы – это вещества, способные в малых количествах замедлять протекание химических процессов или останавливать их. Название ингибитор происходит от латинского inhibere, что означает сдерживать, останавливать. Ещё по данным 1980 года, число известных науке ингибиторов составило более пяти тысяч. Ингибиторы дают народному хозяйству немалую экономию.
Ингибирующее воздействие на металлы, прежде всего на сталь, оказывает целый ряд неорганических и органических веществ, которые часто добавляются в среду, вызывающую коррозию. Ингибиторы имеют свойство создавать на поверхности металла очень тонкую пленку, защищающую металл от коррозии.
Ингибиторы в соответствии с Х. Фишером можно сгруппировать следующим образом.
1) Экранирующие, то есть покрывающие поверхность металла тонкой пленкой. Пленка образуется в результате поверхностной адсорбции. При воздействии физических ингибиторов химических реакций не происходит
2) Окислители (пассиваторы) типа хроматов, вызывающие образование на поверхности металла плотно прилегающего защитного слоя окисей, которые замедляют протекание анодного процесса. Эти слои не очень стойки и при определенных условиях могут подвергаться восстановлению. Эффективность пассиваторов зависит от толщины образующегося защитного слоя и его проводимости;
3) Катодные – повышающие перенапряжение катодного процесса. Они замедляют коррозию в растворах неокисляющих кислот. К таким ингибиторам относятся соли или окислы мышьяка и висмута.
Эффективность действия ингибиторов зависит в основном от условий среды, поэтому универсальных ингибиторов нет. Для их выбора требуется проведение исследований и испытаний.
Наиболее часто применяются следующие ингибиторы: нитрит натрия, добавляемый, например, к холодильным соляным растворам, фосфаты и силикаты натрия, бихромат натрия, различные органические амины, сульфоокись бензила, крахмал, танин и т. п. Поскольку ингибиторы со временем расходуются, они должны добавляться в агрессивную среду периодически. Количество ингибитора, добавляемого в агрессивные среды, невелико. Например, нитрита натрия добавляют в воду в количестве 0,01-0,05%.
Ингибиторы подбираются в зависимости от кислого или щелочного характера среды. Например, часто применяемый в качестве ингибитора нитрит натрия может использоваться в основном в щелочной среде и перестает быть эффективным даже в слабокислых средах.
Применение противокоррозионных
защитных покрытий
Для защиты оборудования и строительных конструкций от коррозии в отечественной и зарубежной противокоррозионной технике применяется большой ассортимент различных химически стойких материалов – листовые и пленочные полимерные материалы, бипластмассы, стеклопластики, углеграфитовые, керамические и другие неметаллические химически стойкие материалы.
В настоящее время расширяется применение полимерных материалов, благодаря их ценным физико-химическим показателям, меньшему удельному весу и др.
Большой интерес для применения в противокоррозионной технике представляет новый химически стойкий материал – шлакоситалл.
Значительные запасы и дешевизна исходного сырья – металлургических шлаков – обусловливают экономическую эффективность производства и применения шлакоситалла.
Шлакоситалл по физико-механическим показателям и химической стойкости не уступает основным кислотоупорным материалам (керамике, каменному литью), широко применяемым в противокоррозионной технике.
Среди многочисленных полимерных материалов, применяемых за рубежом в противокоррозионной технике, значительное место занимают конструкционные пластмассы, а также стеклопластики, получаемые на основе различных синтетических смол и стекловолокнистых наполнителей.
В настоящее время химическая промышленность выпускает значительный ассортимент материалов, обладающих высокой стойкостью к действию различных агрессивных сред. Особое место среди этих материалов занимает полиэтилен. Он инертен во многих кислотах, щелочах и растворителях, теплостоек до температуры + 7000 С и так далее.
Другими направлениями использования полиэтилена в качестве химически стойкого материала являются порошкообразное напыление и дублирование полиэтилена стеклотканью. Широкое применение полиэтиленовых покрытий объясняется тем, что они, будучи одними из самых дешевых, образуют покрытия с хорошими защитными свойствами. Покрытия легко наносятся на поверхность различными способами, в том числе пневматическим и электростатическим распылением.
Также в противокоррозионной технике особого внимания заслуживают монолитные полы на основе синтетических смол. Высокая механическая прочность, химическая стойкость, декоративный вид - все эти положительные качества делают монолитные полы чрезвычайно перспективными.
Продукция лакокрасочной промышленности находит применение в различных отраслях промышленности и строительства в качестве химически стойких покрытий. Лакокрасочное пленочное покрытие, состоящее из последовательно наносимых на поверхность слоев грунтовки, эмали и лака, применяют для противокоррозионной защиты конструкций зданий и сооружений (ферм, ригелей, балок, колонн, стеновых панелей), а также наружных и внутренних поверхностей емкостного технологического оборудования, трубопроводов, газоходов, воздуховодов вентиляционных систем, которые в процессе эксплуатации не подвергаются механическим воздействиям твердых частиц, входящих в состав среды.
В последнее время большое внимание уделяется получению и применению комбинированных покрытий, поскольку в ряде случаев использование традиционных методов защиты является неэкономичным. В качестве комбинированных покрытий, как правило, используется цинковое покрытие с последующей окраской. При этом цинковое покрытие играет роль грунтовки.
Перспективно применение резин на основе бутилкаучука, которые отличаются от резин на других основах повышенной химической стойкостью в кислотах и щелочах, включая концентрированную азотную и серную кислоты. Высокая химическая стойкость резин на основе бутилкаучука позволяет более широко применять их при защите химической аппаратуры.
Данные способы находят широкое применение в промышленности в силу многих своих преимуществ – уменьшения потерь материалов, увеличения толщины покрытия, наносимого за один слой, уменьшения расхода растворителей, улучшение условий производства окрасочных работ и т.д.
38.Электрохимические процессы. Электролиз расплавов и растворов электролитов. Инертные и растворимые электроды. Законы Фарадея
Электрохимические процессы, как и окислительно-восстановительные реакции (ОВР), связаны с изменением степени окисления веществ, участвующих в реакции. Основное отличие ОВР от электрохимических процессов заключается в том, что процессы восстановления и окисления пространственно разделены и перенос электронов может быть зафиксирован как некоторый ток (в гальваническом элементе, при коррозии) или, наоборот, электрохимический процесс может происходить за счет внешнего источника тока (электролиз).
В любом случае для протекания электрохимической реакции необходима электрохимическая цепь, существенными компонентами которой являются электроды и электролит (водный или неводный).
Под электродами обычно понимают или собственно некий проводник или систему, состоящую из проводника, погруженного в раствор электролита. При контакте металлического проводника с раствором электролита на его поверхности возникает некий заряд, за счет переноса электронов, что приводит к возникновению разности электростатических потенциалов между электродом и находящимся с ним в контакте электролитом. Эта разность называется электродным потенциалом.
Абсолютную величину электродного потенциала отдельного электрода измерить невозможно, поэтому измеряют всегда разность потенциалов исследуемого электрода и некоторого стандартного электрода сравнения, т.е. составляют электрохимическую цепь. В качестве электродов сравнения для водных сред используют хлорсеребряный или обратимый водородный электрод сравнения. Последний представляет собой платинированную (электрохимическим способом осажденную на платиновую пластину) платину, погруженную в раствор кислоты (серной, соляной) с активностью ионов водорода равной 1, через который продувают водород при давлении 101кПа. В системе устанавливается равновесие
![]()
![]()
![]() H2(г)
H2(Pt ) 2H(Pt)
2H+ +2e(Pt)
H2(г)
H2(Pt ) 2H(Pt)
2H+ +2e(Pt)
Потенциал этого равновесия в указанных условиях принят равным нулю при любых температурах.
ЭЛЕКТРОЛИЗ РАСПЛАВОВ ЭЛЕКТРОЛИТОВ
Рассмотрим электродные реакции на примере электролиза расплава хлорида натрия.
Под действием температуры ионная кристаллическая решетка NaCl разрушается на ионы Na+ и Cl-. Если погрузить в расплавленную соль два графитовых (инертных) электрода и подключить их к полюсам внешнего источника тока, то в электролите начнется направленное движение ионов и на электродах будут происходить следующие реакции:
а) восстановление ионов Na+ до металлического натрия (катодный процесс).
б) окисление ионов Cl- до газообразного хлора (анодный процесс).
(-) K NaCl A (+)
![]()
![]()
Na+ + Cl- ®
Суммируя уравнения катодного и анодного процессов, (с учетом электронного баланса) получим окислительно-восстановительную реакцию, протекающую при электролизе:
2NaCl = 2Na(ж) + Cl2(г)
ЭЛЕКТРОЛИЗ РАСТВОРОВ ЭЛЕКТРОЛИТОВ
При электролизе растворов электролитов происходит конкуренция между растворенным веществом и растворителем за участие в электродных процессах. Например, в водных растворах солей кроме анионов и катионов соли всегда имеются молекулы H2O и ионы H+ и OH-. При наличии нескольких видов ионов или недиссоциированных молекул электрохимически активных веществ возможно протекание нескольких электродных реакций.
Для объяснения электродных процессов, происходящих при электролизе разбавленных водных растворов электролитов, можно руководствоваться следующими правилами:
На катоде в первую очередь восстанавливаются (принимают электроны) катионы с наиболее высокими (максимальными) значениями электродного потенциала (сильные окислители).
Если электродный потенциал катиона j°Me/Men+ < -1,6 В, то на катоде восстанавливаются ионы H+ молекулы H2O с выделением H2 и
накоплением в растворе OH-:
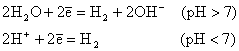
При
значении
стандартного
электродного
потенциала
катиона
![]() на катоде
восстанавливаются
только ионы
металла, а разряд
ионов H+ не происходит:
на катоде
восстанавливаются
только ионы
металла, а разряд
ионов H+ не происходит:
![]()
Если -1,6 В <j°Me/Men+ < 0 В, то на катоде протекают два процесса: восстановление ионов Н+ и ионов металла. Это обусловлено тем, что, во-первых, потенциал водородного электрода зависит от рН раствора:
jH2/2H+ = -0,059· pH,
во-вторых, выделение водорода на катоде происходит с более высоким перенапряжением по сравнению с перенапряжением разряда многих металлов.
Таким образом, при некоторой плотности тока потенциал выделения водорода становится отрицательнее, чем потенциал выделения металла (рис.8.15).
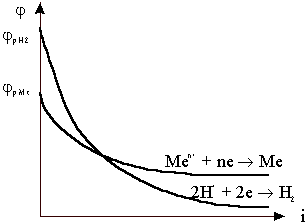
Высокое водородное перенапряжение - явление положительное, благодаря этому, из водных растворов удается выделять на катоде марганец, цинк, хром, железо, кадмий, кобальт, никель и другие металлы. Все вышесказанное можно представить следующей схемой:
ЗАКОНЫ ЭЛЕКТРОЛИЗА
Количественные соотношения при электролизе были исследованы английским физиком М. Фарадеем и описаны двумя законами.
Первый закон Фарадея. Масса веществ, выделяющихся на электродах, прямо пропорциональна количеству электричества, прошедшего через электролит.
m = k· Q, где
Q - количество электричества (Кл),
k - константа, электрохимический эквивалент.
Так как Q = I· t, то m = k· I· t, где
I - сила тока в амперах (А);
t - продолжительность электролиза в секундах (с).
Второй закон Фарадея. Равные количества электричества выделяют при электролизе из различных электролитов эквивалентные количества вещества.
nэк.(A) = nэк.(B) или
![]()
Для восстановления на катоде и окисления на аноде 1 моль эквивалентов вещества через электролит должно пройти 96500 кулонов электричества. Это количество электричества называют числом Фарадея (F).
F = I· t = 96500 Кл/моль.
Оба закона можно свести в одну формулу:
![]() где m - масса
вещества (г);
где m - масса
вещества (г);
Mэк.
- молярная масса
эквивалентов
выделившегося
вещества (г/моль);
![]() где M - молярная
масса выделившегося
вещества (г/моль);
где M - молярная
масса выделившегося
вещества (г/моль);
Z - число эквивалентности, равное числу электронов, участвующих в процессе окисления или восстановления на электродах при получении 1 моль вещества.
Если необходимо рассчитать объем газа, выделившегося в процессе электролиза, то выражение законов Фарадея может быть записано следующим образом
![]()
где V° - объем выделившегося газа при н.у., (л);
Vэк. - молярный объем эквивалентов газа (л/моль);
![]()
где Vm - молярный объем газа, равный 22.4 л/моль.
В случае параллельных процессов часть количества электричества расходуется на выделение одного вещества, часть - на выделение другого. Доля общего количества электричества (в процентах), которая расходуется на выделение одного из веществ, называется выходом по току этого вещества:
![]()
Где ВТ - выход по току вещества А;
QА - количество электричества, израсходованное на превращение вещества А;
Q - общее количество электричества, прошедшее через электрод.
Так как по первому закону Фарадея
m = k· Q, то
![]()
Где mА - масса реально выделенного вещества А на электроде;
m - теоретическая масса вещества А, рассчитанная по закону Фарадея.
ПРИМЕНЕНИЕ ЭЛЕКТРОЛИЗА
Электролиз широко используется в различных областях народного хозяйства.
В энергетике водород, полученный электролизом, используют для охлаждения генераторов на тепловых и атомных электростанциях.
Электролизом растворов солей получают медь, цинк, кадмий, никель, кобальт, марганец и другие металлы. В этих процессах используют нерастворимые аноды.
Электролизом расплавов соединений получают алюминий, магний, натрий, литий, бериллий и кальций, а также сплавы некоторых металлов.
Метод электролиза используют для рафинирования (очистки) металлов: меди, золота, серебра, свинца, олова и др. При рафинировании анодом служит очищаемый металл. На аноде растворяется основной металл и примеси, потенциал которых отрицательнее потенциала основного металла. Примеси, имеющие более положительный потенциал, выпадают из анода в виде шлама.
Электролиз используется для нанесения металлических покрытий на металлы и пластмассы (гальванические покрытия). При этом катодом служит обрабатываемое изделие, анодом - или металл покрытия, или нерастворимый электрод.
Электроды в электрохимии, электронно-проводящие фазы, контактирующие с ионным проводником (электролитом). Часто под электродами понимают лишь одну электронно-проводящую фазу. При пропускании тока от внешнего источника через систему из двух электродов, соединенных друг с другом через электролит, на электроды протекают два процесса: заряжение двойного электрического слоя и электрохимическая реакция. В отличие от фазовых контактов металл-металл, металл-полупроводник, полупроводник-полупроводник и т. п. на границе фаз, составляющих электрохимическую систему, вид носителей тока меняется, т. к. в электролите ток переносится ионами, а в электронно-проводящей фазе - электронами, Непрерывность прохождения тока через границу фаз в этом случае обеспечивается электродной реакцией. Электрод называют анодом, если на его поверхности преобладает реакция, приводящая к генерированию электронов, т. е. происходит окисление веществ, содержащихся в электролите, либо ионизация металла анода. Электрод называют катодом, если с его поверхности электроны металла переходят на частицы реагирующих веществ, которые при этом восстанавливаются.
Классификация электродов проводится по природе окислителей и восстановителей, которые участвуют в электродном процессе. Электродом 1-го рода называют металл (или неметалл), погруженный в электролит, содержащий ионы этого же элемента. Металл электродов является восстановленной формой вещества, а его окисленной формой - простые или комплексные ионы этого же металла. Например, для системы Сu Сu2+ + 2е, где е - электрон, восстановленной формой является Сu, а окисленной - ионы Сu2+, Соответствующее такому электродному процессу Нернста уравнение для электродного потенциала Е имеет вид:
![]()
где
E° - стандартный
потенциал при
т-ре Т;
![]() термодинамическая
активность
ионов Сu2+; F - постоянная
Фарадея, R - газовая
постоянная,
К электродам
1-го рода относятся
амальгамные
электроды, так
как для них
восстановленная
форма - амальгама
металла, а окисленная
- ионы этого же
металла. Например,
для амальгамы
таллия устанавливается
равновесие:
Tl+ + e(Hg)
термодинамическая
активность
ионов Сu2+; F - постоянная
Фарадея, R - газовая
постоянная,
К электродам
1-го рода относятся
амальгамные
электроды, так
как для них
восстановленная
форма - амальгама
металла, а окисленная
- ионы этого же
металла. Например,
для амальгамы
таллия устанавливается
равновесие:
Tl+ + e(Hg)
![]() Tl(Hg). В такой
системе могут
изменяться
концентрации
и окисленной,
и восстановленной
форм, поэтому
уравнение
Нернста имеет
вид:
Tl(Hg). В такой
системе могут
изменяться
концентрации
и окисленной,
и восстановленной
форм, поэтому
уравнение
Нернста имеет
вид:
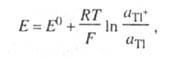
где aтl, - термодинамическая активность таллия в амальгаме.
Электроды
2-го рода- системы
из металла М,
покрытого слоем
его же труднорастворимой
соли
![]() или оксида,
и погруженного
в раствор, содержащий
анионы этой
соли (для оксида
- ионы ОН-). Окисленной
формой является
соль
или оксида,
и погруженного
в раствор, содержащий
анионы этой
соли (для оксида
- ионы ОН-). Окисленной
формой является
соль
![]() а восстановленная
форма представлена
металлом М и
анионом Az-:
а восстановленная
форма представлена
металлом М и
анионом Az-:
![]() где
где
![]() зарядовое число
иона. В системе
устанавливается
равновесие
между атомами
М и анионами
Az-в растворе,
которое включает
два "парциальных"
равновесия:
между металлом
и катионом соли
и между анионом
соли в ее твердой
фазе и анионом
в растворе.
Такие электроды
называют обратимыми
по аниону.
зарядовое число
иона. В системе
устанавливается
равновесие
между атомами
М и анионами
Az-в растворе,
которое включает
два "парциальных"
равновесия:
между металлом
и катионом соли
и между анионом
соли в ее твердой
фазе и анионом
в растворе.
Такие электроды
называют обратимыми
по аниону.
Уравнение Нернста имеет вид:
![]()
К электроды 2-го рода относятся многие электроды сравнения, например, каломельный, хлорсеребряный, оксидно-ртутный.
Электроды 3-го рода - системы из металла, контактирующего с двумя труднорастворимыми солями. В результате электрохимической реакции на электроде менее растворимая соль превращается в более растворимую, а потенциал электроды определяется термодинамической активностью катионов более растворимой соли. Так, в системе Рb2+| РbС12, AgCl, процесс Ag происходит
![]()
Металл электрода может не участвовать в реакциях, а служить лишь передатчиком электронов от восстановленной формы вещества к окисленной; такие электроды называют окислительно-восстановительными или редокс-электродами. Например, платиновый электрод в растворе, содержащем ионы [Fe(CN)6]4- и [Fe(CN)6]3-, осуществляет перенос электронов между этими ионами в качестве передатчика (медиатора). Среди окислительно-восстановительных электродов выделяют газовые электроды, состоящие из химически инертного металла (обычно Pt), к которому подводится электрохимически активный газ (напр., Н2 или Сl2). Молекулы газа адсорбируются на поверхности металла, распадаясь на адсорбированные атомы, которые непосредственно участвуют в переносе электронов через границу раздела фаз. Наиболее распространен водородный электроды, на поверхности которого образуются адсорбированные атомы Надс и устанавливается равновесие: Н2 2Надс 2Н+ + 2е. Разл. типы электроды можно объединить в рамках так называемой концепции электронного равновесия на границе металл-электролит, согласно которой каждому равновесному электродному потенциалу соответствует определенная термодинамическая активность электронов в электролит.
электроды называют идеально поляризуемым, если вследствие термодинамических или кинетических причин переход электронов через межфазную границу невозможен. При изменении потенциала такого электрода происходит только изменение строения двойного электрического слоя, что сопровождается протеканием тока заряжения, спадающего до нуля, когда перестройка двойного электрического слоя заканчивается. Для неполяризуемых, или обратимых, электродов переход электронов через границу фаз, напротив, незаторможен, и при пропускании тока через такой электроды его потенциал практически не изменяется.
По функциям в электрохимической системе электроды подразделяют на рабочие, вспомогательные и электроды сравнения. Рабочим называют электрод, на котором происходит исследуемый электрохимический процесс. Вспомогательный электрод (или противоэлектрод) обеспечивает возможность пропускания тока через электрохимическую ячейку, а электроды сравнения - возможность измерения потенциала рабочего электроды Специфика широко используемых в электрохимии жидких электродов (ртуть, амальгамы, галлий, жидкие сплавы на основе Ga - галламы, расплавы металлов и т. п.) связана с идеальной гладкостью их поверхности, истинная площадь которой совпадает с ее геометрической величиной, а также с энергетической однородностью и изотропностью свойств поверхности электроды и возможностью растворения выделяющихся металлов в материале электрода.
На практике электроды классифицируют по химической природе материала (металлические, неметаллические, оксидные, электроды из соединений с ковалентной связью, углеграфитовые и т.д.), форме (сферические, плоские, цилиндрические, дисковые и т. д.), условиям функционирования (неподвижные, вращающиеся и т. п.), размерам (микро- и ультрамикроэлектроды), пористости, гидрофильности, участию электродного материала в электродном процессе (расходуемые и нерасходуемые) и др. признакам. Использование капельного ртутного электрода лежит в основе полярографии. Вращающийся дисковый электродпредставляет интерес как система, для которой существует строгое решение диффузионной кинетической задачи. К особо практически важным электродам следует отнести каталитически активные и высоко коррозионностойкие оксидные рутениево-титановые аноды (ОРТА), применение которых революционизировало самое широкомасштабное электрохимическое производство - электролитическое получение хлора и щелочей.
Модифицирование электродов, получившее широкое распространение в электрокатализе, производстве химических источников тока, электрохимических сенсоров и т. п., основано как на физических (ионная имплантация, разрыхление поверхности, выращивание монокристаллических граней, создание монокристаллических структур, физическая адсорбция ионов и молекул и др.), так и химических методах. В частности, химически модифицированные электроды представляют собой проводящий или полупроводниковый материал, покрытый мономолекулярными (в т. ч. субатомными), полимолекулярными, ионными, полимерными слоями, в результате чего электроды проявляет химические, электрохимические и/или оптические свойства слоя. Химическое модифицирование достигается хемосорбцией на поверхности электроды ионов и молекул, ковалентным связыванием различных агентов с поверхностными атомными группами, покрытием поверхности органическими, металлорганическими или неорганическими полимерными слоями, созданием композитов из электродного материала и вещества - модификатора.
Микроэлектроды имеют по крайней мере один из размеров настолько малый, что свойства электроды оказываются размернозависимыми. Размеры микроэлектродов лежат в интервале 0,1-50 мкм, минимальная площадь составляет 10-14 м2 (ультрамикроэлектроды), тогда как в большинстве электроаналитических экспериментов применяют электроды с площадью 5 х 10-5м2, в лабораторном электросинтезе - 10-2 м2, Осн. преимущество микроэлектродов - возможность снизить с их помощью диффузионные ограничения скорости электродного процесса и, следовательно, изучать кинетику очень быстрых электродных реакций. Из-за малой величины токов электрохимической ячейки с микроэлектродами характеризуются незначительным омическим падением потенциала, что позволяет изучать системы с высокими концентрациями реагирующих частиц, обычно используемые в технологических процессах, применять высокие скорости сканирования потенциала при вольтамперометрических измерениях, проводить работы в плохо проводящих средах и т. п. Микроэлектроды используют для анализа ультрамалых проб, исследования процессов в живых организмах, в клинических целях. Ультрамикроэлектроды применяют в туннельной сканирующей микроскопии и в электрохимической нанотехнологии.
39. Распространенность химических элементов. Основные классы неорганических соединений
РАСПРОСТРАНЁННОСТЬ ЭЛЕМЕНТОВ
относительное содержание элементов в космич. веществе. Часто под Р. э. подразумевают распространённость не только хим. элементов, но также и их изотопов по отдельности, т. е. более общее понятие - распространённость нуклидов (РН). Среднюю РН определяют по совокупности данных геохимии, космохимии и астрофизики тремя осн. методами: исследованием состава образцов земного, метеоритного и лунного вещества; изучением спектров эл.-магн. излучения Солнца, звёзд и межзвёздной среды; определением содержания нуклидов в солнечных и галактич. космических лучах.
Рис.
1. Относительная
распространённость
нуклидов lgN (N-
число атомов,
IgNSi = 6) в зависимости
от атомной
массы А (по А.
Камерону). Изотопы
одного и того
же элемента
(вплоть до Ge)
соединены
прямыми линиями.
Символы указывают
основные процессы
синтеза нуклидов:
D - взрывное горение
С, О и Si, О - медленный
захват нейтронов
(s-процесс), + - быстрый
захват нейтронов
(r -процесс),
![]() сравнимый
вклад s- и r -процессов,
0 - ядерное статистическое
равновесие
( е -процесс).
Нуклиды, образующиеся
в других процессах,
отмечены точками.
Штриховой
линией соединены
обойдённые
ядра.
сравнимый
вклад s- и r -процессов,
0 - ядерное статистическое
равновесие
( е -процесс).
Нуклиды, образующиеся
в других процессах,
отмечены точками.
Штриховой
линией соединены
обойдённые
ядра.
Изотопный состав вещества достаточно хорошо изучен только для Солнечной системы. В Солнце заключена б. ч. массы Солнечной системы. Однако спектральный анализ содержания элементов и нуклидов в солнечной атмосфере не обладает столь большой точностью, как хим., радиохим. и масс-спектроскопич. анализы состава метеоритного и планетного твёрдых веществ. Поэтому содержание нуклидов в метеоритах рассматривается в качестве стандарта при систематизации распространённости большинства элементов.
На рис. 1 в логарифмич. шкале показана РН в Солнечной системе, нормированная на содержание кремния. Приведённые данные получены в осн. из анализа состава метеоритов. Систематизация этих данных выполнена А. Камероном (A. Cameron) в 1982 (см. также табл.). Наиб. распространённость имеет водород (1 Н), примерно на порядок меньше - гелий (4 Не). Т. к. распространённость этих элементов вследствие их летучести на Земле, Луне и метеоритах мала, их действит. содержание в природе оценивают с привлечением косвенных данных: анализа внутр. строения звёзд и состава вещества межзвёздной среды, а также выводов космологии. Водород и гелий имеют в осн. первичное, космологич. происхождение (см. Горячей Вселенной теория). Низкое содержание дейтерия и изотопов Li, Be, В объясняется тем, что эти нуклиды при звёздных темп-pax легко вступают в разл. ядерные реакции.
РН в ср. быстро падает с увеличением массового числа, обнаруживая максимумы для групп С, N, О и Fe ("железный пик") и затем неск. двойных пиков, соответствующих элементам Кг и Sr, Хе и Ва, Pt и Pb, к-рые имеют устойчивые изотопы с магич. числами нейтронов 50, 82, 126 (см. Магические ядра )либо получаются при бета-распаде ядер с такими нейтронными числами.
На рис. 2 та же кривая РН приведена в более компактном виде, без разделения изотопов по процессам их образования. Эта т. н. стандартная кривая РН в Солнечной системе, построенная согласно данным А. Камерона, чётко обнаруживает указанные выше максимумы и является гл. наблюдат. основой теории нуклеосинтеза в природе. Согласно этой теории, осн. процессы образования ядер в природе включают космологич. нуклеосинтез в горячей Вселенной, приводящий к образованию гелия, термоядерное горение лёгких элементов от водорода до кремния в недрах звёзд, синтезирующее элементы "железного пика", а также процессы медленного и быстрого захвата нейтронов ядрами с образованием тяжёлых нуклидов вплоть до изотопов висмута и урана. Особый интерес в теории нуклеосинтеза представляет происхождение т. н. обойдённых ядер. Это изотопы Se, Mo, Cd, La, Dy и др. элементов, к-рые оказываются в стороне от путей нейтронного захвата. Распространённость обойдённых нуклидов примерно на два порядка меньше распространённости ядер, образующихся в процессах нейтронного захвата. Синтез обойдённых ядер объясняют обычно ядерными реакциями с участием протонов (р, у),(r, h) или слабыми взаимодействиями с участием нейтрино, возникающими при взрыве сверхновой. Не исключён также вклад в механизм их синтеза тройного деления ядер с вылетом обогащённых нейтронами лёгких за-ряж. частиц.
рис 2
Несмотря на то, что состав большинства звёзд, галактик и межзвёздной среды в осн. следует стандартной кривой РН, существуют отклонения от неё, вызванные разл. физ. причинами. Старые звёзды, принадлежащие гало Галактики и шаровым звёздным скоплениям, содержат тяжёлых элементов в 10-103 раз меньше, чем Солнечная система. Это связано с хим. эволюцией галактик. Нек-рые группы звёзд содержат тяжёлые элементы в пропорциях, существенно отличающихся от стандартных распространённостей, таковы, напр., т. н. суперметаллич. звёзды (бариевые, CNO и др.). Существуют также обогащённые и обеднённые гелием звёзды, звёзды с низким содержанием Са. Звёзды с аномальным хим. составом составляют примерно 10% всех звёзд, находящихся вблизи гл. последовательности (см. Герцшпрунга - Ресселла диаграмма )и имеющих темп-ру поверхности от 8000 до 20 000 К (см. Химически пекулярные звёзды).
Появились свидетельства в пользу того, что изотопный состав Солнечной системы также не является столь однородным, как казалось раньше. Открыты аномалии (большинство из них на уровне долей процента) в рас-пространённостях изотопов кислорода, неона, магния. Всё это указывает на многообразие процессов, сформировавших вещество звёзд, галактик и Солнечной системы. Происхождение и распространенность химических элементов в природе
Вам хорошо известно, что различные химические элементы распространены крайне неравномерно. Элемент может быть в сотни и тысячи раз более или менее распространенным, чем его непосредственный сосед по периодической системе. Вы знаете, что атомов одних элементов (кислород, кремний, алюминий, железо и др.) на нашей планете значительно больше, чем атомов других элементов (медь, золото, германий и др.). А откуда вообще взялось такое разнообразие химических элементов? Давайте, прежде чем перейти к рассмотрению вопроса об относительной распространенности химических элементов, кратко познакомимся с существующей точкой зрения по вопросу их происхождения.
По принятой сейчас модели развития Вселенной, формирование слагающего ее вещества является результатом «Большого взрыва». В первые мгновения после него произошло формирование элементарных частиц. Вначале – фотонов, нейтрино, электронов, позитронов. Затем – протонов и нейтронов. После снижения температур ниже уровня 1011о К начинается соединение протонов с нейтронами. Образуются ядра тяжёлых изотопов водорода, возможно также ядер гелия, и небольших количеств Li, Be.
Синтез более тяжёлых атомных ядер начинается после формирования крупных и плотных горячих газовых скоплений – звёзд. Вначале – продолжается образование 4Не. Далее же происходит т.н. «выгорание» гелия:
34Не Ю12С
и далее, с присоединением новых ядер гелия: 16O, 20Ne, 24Mg, 28Si, 32S и т.д., вплоть до 56Fe и 58Ni. Обратите внимание, что всё это – именно синтез ядер (нуклеосинтез), а не атомов в целом, так как электроны при столь высоких температурах остаются в свободном состоянии.
Образование ядер промежуточных элементов – результат реакций захвата либо потери протона или нейтрона.
Атомы тяжелее Fe и Ni в обычных процессах внутризвёздного нуклеосинтеза не формируются (не хватает энергии). Эти процессы реализуются только при взрывах «сверхновых» звезд. При наблюдении за сверхновыми в их спектре обнаружены яркие линии, характерные для 254Cf. Интересно, что скорость падения яркости сверхновых (56 суток) очень точно совпадает с периодом полураспада калифорния. Таким образом, формирование ядер атомов от никеля до урана – результат ядерного синтеза в процессе взрыва сверхновых, а также распада калифорния и, возможно, других трансурановых элементов (может, и более тяжёлых, которые нам неизвестны).
Существуют звёзды первого и второго поколения. Только вторые могут содержать в составе элементы тяжелее никеля и иметь планетные системы типа Солнечной.
Итак, в химическом отношении звезды являются довольно простыми системами. Доступная для изучения часть Вселенной имеет в основном водородно-гелиевый состав. Сбылось предсказание английского астрофизика А. Эддингтона, который в начале ХХ века писал, что легче будет разобраться в составе звезд, чем в процессах, окружающих нас на Земле.
Закономерности распространения химических элементов в космосе и на Земле вначале были установлены чисто эмпирически. Было подмечено, что:
Распространенность быстро падает от элементов с низкими атомными номерами (примерно до номера 30), а затем, для более тяжелых элементов остается приблизительно постоянной.
Только десять элементов – H, He, C, N, O, Ne, Mg, Si, S, Fe, атомные номера которых меньше 27, характеризуются высокой распространенностью; из них водород резко преобладает над остальными.
Элементы с четными порядковыми номерами более распространены, чем нечетные (закон Оддо - Гаркинса).
Уточнения к закону Оддо-Гаркинса впоследствии сформулировали А.Е. Ферсман и другие геохимики, но основная суть его остаётся неизменной. Истоки закономерностей – в строении атомных ядер. Первоначально геохимики предполагали, что это может быть как-то связано с различной степенью устойчивости атомных ядер различных элементов. Сейчас признаётся, что это отражает механизм термоядерного синтеза в космических условиях.
Установленные закономерности показывают, что абсолютная распространенность элементов зависит в большей степени от свойств ядра, чем от химических свойств элемента и связана со стабильностью ядер.
А.Е. Ферсман заметил, что все химические элементы можно подразделить на 4 группы с порядковыми номерами, выражающимися формулами:
4q 4q+3 4q+2 4q+1,
которые составляют 86,19%,12,74%, 0,05%- и 0,02% по массе соответственно
Элемент однозначно характеризуется числом протонов в ядре, но число нейтронов может колебаться. В результате элемент может иметь несколько изотопов, различающихся по массовому числу или атомному весу и стабильностью, но практически неотличимых по химическим свойствам. С другой стороны, существуют изобары, которые являются разными элементами, но имеют одинаковое число нейтронов.
Ядра, содержащие 2, 8, 20, 28, 50, 82, 126 протонов или нейтронов особенно устойчивы. Эти числа называются магическими. Наиболее устойчивы дважды магические ядра, содержащие магическое число и протонов и нейтронов – 4He, 16O, 40Ca. В земной коре элементы с магическими ядрами обладают достаточно высокой распространенностью (за исключением гелия).
Обобщая все данные о распространённости химических элементов и их поведении в геохимических процессах, В.М. Гольдшмидт сформулировал основной закон геохимии:
Содержания химических элементов зависят от строения их атомного ядра, а их миграция – от строения электронных оболочек, определяющих химические свойства элементов. Для геохимии в равной мере важны оба этих аспекта.
Одним из основных законов геохимии является закон Ферсмана-Гольдшмидта, который можно сформулировать следующим образом: Геохимия элемента в земной коре определяется как химическими свойствами, так и величиной кларка.
Основные классы неорганических соединений
Неорганические вещества классифицируются по составу и по химическим свойствам. По составу неорганические вещества делятся на бинарные – состоящие только из двух элементов, и многоэлементные – состоящие из нескольких элементов. Бинарные соединения классифицируются по неметаллу, например CaH2, NaH – гидриды, CaS, FeS – сульфиды, СаС2, Al4C3 – карбиды и т. д. Многоэлементные соединения классифицируются по общему элементу, чаще всего кислороду, например: NaNO3, H2SO4, KClO4 – кислородсодержащие.
Далее будут рассмотрены четыре важнейших класса неорганических соединений: оксиды, гидроксиды металлов, (гидроксиды неметаллов относятся, как правило, к кислотам) кислоты, соли.
Оксиды
Оксидами называются бинарные соединения, содержащие кислород в степени окисления -2.
По химическим свойствам оксиды делятся на солеобразующие и несолеобразующие. Солеобразующие, в свою очередь, делятся на основные, кислотные и амфотерные.
Основные оксиды взаимодействуют с кислотами с образованием соли и воды, например:
CuO + 2HCl = CuCl2 + H2O
MnO + H2SO4 = MnSO4 + H2O
В состав основных оксидов входят металлы главных подргрупп I и II групп Периодической системы (кроме бериллия), а также переходные металлы в низших степенях окисления, например СаО, К2О, MnO, FeO, CrO.
Основные оксиды, образованные щелочными и щелочноземельными металлами взаимодействуют с водой с образованием щелочей:
Na2O + H2O = 2NaOH
CaO + H2O = Ca(OH)2
Кислотными оксидами называются оксиды, взаимодействующие со щелочами с образованием соли и воды, например:
SO2 + 2KOH = K2SO3 + H2O
CO2 + 2NaOH = Na2CO3 + H2O
В состав кислотных оксидов входят неметаллы или переходные металлы в высших степенях окисления, например: P2O5, SiO2, CrO3, Mn2O7.
Кислотные оксиды (кроме SiO2) взаимодействуют с водой:
SO3 + H2O = H2SO4
P2O5 + 3H2O = 2H3PO4
Амфотерные оксиды в зависимости от условий проявляют свойства основных или кислотных оксидов, т.е. образуют соли как с кислотами, так и с основаниями, например:
Cr2O3 + 6HCl = 2CrCl3 + 3H2O
Cr2O3 + 2NaOH = 2NaCrO2 + H2O
В состав амфотерных оксидов входят переходные металлы в промежуточных степенях окисления, металлы главной подгруппы III группы, например Cr2O3, Al2O3, MnO2. К амфотерным оксидам относятся также BeO, ZnO и PbO2. Амфотерные оксиды с водой не взаимодействуют.
Несолеобразующие оксиды не дают реакций, характерных для солеобразующих оксидов. К ним относятся: NO, N2O, SiO, CO. Несолеобразующие оксиды могут реагировать с кислотами или щелочами, но при этом не образуются продукты, характерные для солеобразующих оксидов, например при 150oС и 1,5 Мпа СО реагирует с гидроксидом натрия с образованием соли – формиата натрия:
СО + NaOH = HCOONa
Однако вода в этой реакции никогда не образуется, поэтому СО относят к несолеобразующим оксидам.
Оксиды можно получить следующими основными способами:
1. из простых веществ:
2Cu + O2 = CuO
S + O2 = SO2
2. окислением сложных веществ:
4FeS2 + 11O2 = 2Fe2O3 + 8SO2
CH4 + 2O2 = CO2 + 2H2O
6MnO + O2 = 2Mn3O4
3. термическим разложением оксидов, гидроксидов, кислородсодержащих солей и кислот:
3MnO2 = Mn3O4 + O2
2Fe(OH)3 = Fe2O3 + 3H2O
2Cu(NO3)2 = 2CuO + 4NO2 + O2
H2SiO3 = SiO2 + H2O
Гидроксиды
Гидроксидами металлов называются вещества, содержащие ион металла и одну или несколько гидроксильных групп.
Гидроксиды делятся на основные (основания) и амфотерные. Основные гидроксиды, в свою очередь, делятся на сильные основания – щелочи, и слабые основания. В состав щелочей входят катионы щелочных и щелочноземельных металлов, например КОН, NaOH, Ca(OH)2, Ba(OH)2. Слабыми основаниями являются гидроксиды переходных металлов в низших степенях окисления, например Fe(OH)2, Mn(OH)2, Cu(OH)2.
Число гидроксильных групп в основании называется кислотностью основания.
Амфотерные гидроксиды включают в свой состав катионы металлов III группы Периодической системы, катионы переходных металлов в промежуточных степенях окисления, например Al(OH)3, Cr(OH)3, Fe(OH)3. К амфотерным также относятся Be(OH)2, Zn(OH)2.
Основные гидроксиды реагируют с кислотами с образованием соли и воды, например:
Сu(OH)2 + H2SO4 = CuSO4 + 2H2O
Щелочи реагируют с кислотными и амфотерными оксидами:
Ca(OH)2 + CO2 = CaCO3 + H2O
2NaOH + Fe2O3 = 2NaFeO2 + H2O
Амфотерные гидроксиды реагируют и с кислотами (в этом случае они ведут себя как основания), и со щелочами (как кислоты), например:
Al(OH)3 + 3NaOH = Na3[Al(OH)6]
Al(OH)3 + 3HCl = AlCl3 + 3H2O
Cлабые основания и амфотерные гидроксиды при нагревании разлагаются:
Cu(OH)2 = CuO + H2O
2Fe(OH)3 = Fe2O3 + 3H2O
Для получения слабых оснований и амфотерных гидроксидов используют реакцию их вытеснения из солей щелочами:
CuCl2 + 3NaOH = Cu(OH)2 + 3NaCl
Fe2(SO4)3 + 6KOH = Fe(OH)3 + 3K2SO4
Щелочи можно получить взаимодействием металла с водой:
2Na + 2H2O = 2NaOH + H2
соответствующего оксида с водой:
СаО + Н2О = Са(ОН)2
или электролизом водного раствора соли соотвествующего металла:
2KCl + 2H2O = 2KOH + H2 + Cl2
Кислоты
Кислоты реагируют с основаниями (а также с основными и амфотерными оксидами и гидроксидами) с образованием солей. Например:
HCl + NaOH = NaCl + H2O
H2SO4 + Fe(OH)2 = FeSO4 + 2H2O
2HNO3 + ZnO = Zn(NO3)2 + H2O
Кислоты классифицуируются по следующим признакам:
по силе (как электролиты) - на сильные (например HCl, HNO3, H2SO4) и слабые (H2S, HNO2, HCN и т.д.)
по наличию кислорода в составе кислоты - на кислородные (HClO3, H3PO4) и бескислородные (HCN, H2S). При этом элемент, входящий в состав кислородной кислоты называется кислотообразующим.
основности (т.е. по числу атомов водорода в молекуле кислоты, способных замещаться на металл) на одноосновные (HCl, HNO3), двухосновные (H2SO3, H2S), трехосновные (H3PO4) и т.д.
по окислительным свойствам - на обычные кислоты, у которых в окислительно-восстановительных реакциях, например с металлами, восстанавливаются ионы водорода (например, HCl), и кислоты-окислители, у которых происходит восстановление кислотообразующего элемента (например, HNO3).
Кислоты имеют общие химические свойства:
Взаимодейcтвуют с металлами. Обычные кислоты (неокислители) взаимодействуют с металлами, стоящими в ряду напряжений левее водорода:
Fe + 2HCl = FeCl2 + H2
Zn + H2SO4(разб) = ZnSO4 + H2
Кислоты окислители могут реагировать как с металлами, расположенными в ряду напряжений левее водорода, например:
Zn + HNO3(разб) = Zn(NO3)2 + H2O + N2
Так и правее его:
Ag + HNO3(конц) = AgNO3 + H2O + NO2
Соли
Соли можно рассматривать как продукт взаимодействия основания и кислоты. При этом может происходить как полное, так и неполное замещение ионов водорода в кислоте катионами металла (или аммония) или гидроксильных групп в основании кислотными остатками.
Соли, не содержащие ионов водорода или гидроксильных групп, называются средними, например NaCl, CuSO4, Ca3(PO4)2.
Соли, содержащие ионы водорода – кислые, например: KH2PO4 – дигидрофосфат калия, NaHCO3 – гидрокарбонат натрия.
Соли, содержащие ионы гидроксила, называются основными: Mg(OH)Cl – гидроксихлорид магния, (CuOH)2CO3 – гидроксикарбонат меди (II).
Соли, содержащие два катиона, называются двойными: Fe(NH4)2(SO4)212H2O – двойной сульфат железа(II)-аммония (соль мора), KCr(SO4)212H2O – двойной сульфат хрома (III) - калия (хромокалиевые квасцы).
Соли, содержащие комплексные ионы, называются комплексными: K3[Fe(CN)6] - гексацианоферрат (III) калия (красная кровяная соль), [Co(NH3)6]Cl2 - хлорид гесаамминокобальта (II).
Солями называются электролиты, дающие при диссоциации в водном расторе катиона металла или аммония (и водорода в случае кислых солей) и анионы кислотного остатка (и гидроксила в случае основных солей). Ионы, входящие в состав соли могут быть комплексными.
Соли реагируют с металлами, эти реакции всегда окислительно-восстановительные:
Fe + CuSO4 = Cu + FeSO4
Cu + FeCl3 = CuCl + FeCl2
C неметаллами, это также окислительно-восстановительные реакции:
S + Na2SO3 = Na2S2O3 - при кипячении
С водой, образуя кристаллогидраты:
CuSO4 + 5Н2О = CuSO45H2O
Na2SO4 + 10Н2О = Na2SO410H2O
или необратимо гидролизуясь:
Al2S3 + 6Н2O = 2Al(OH)3 + 3H2S
Соли реагируют со щелочами:
NH4Cl + NaOH = NH3 + NaCl + H2O
CuCl 4 + NaOH = NaCl + Cu(OH)2
и кислотами:
K2CO3 + HCl = KCl + CO2 + H2O
NaNO3(тв) + H2SO4(конц) = NaHSO4 + HNO3 - при нагревании
Ca3(PO4)2 + H3PO4 = 3CaHPO4
Соли реагируют с солями:
NaCl + AgNO3 = NaNO3 + AgCl
Соли кислородных кислот при нагревании разлагаются:
2KClO3 = 2KCl + 3O2
2NaNO3 = 2NaNO2 + O2.
40. Общие закономерности химии s-элементов: s-элементы I, II и III групп периодической системы Д.И. Менделеева (физические, химические свойства, способы получения, применение, биологическая роль)
s-блок в периодической таблице элементов — электронная оболочка, включающая в себя первые два слоя s-электронов.
Данный блок включает в себя щелочные металлы, щелочноземельные металлы, водород и гелий.
Эти элементы отличаются тем, что в атомном состоянии высокоэнергичный электрон находится на s-орбитали. Исключая водород и гелий, эти электроны очень легко переходят и формируются в позитивные ионы при химической реакции. Конфигурация гелия химически весьма стабильна, следовательно, именно по этому гелий не имеет стабильных изотопов; иногда, благодаря этому свойству, его объединяют с инертными газами.
Остальные элементы, имеющие этот блок, все без исключения являются сильными восстановителями и поэтому в свободном виде в природе не встречаются. Элемент в металлическом виде может быть получен только с помощью электролиза растворенной в воде соли. Дэви Гемфри, в 1807 и 1808 году, стал первым кто отсоединил соли кислот от s-блок-металлов, за исключением лития, бериллия, рубидия и цезия. Бериллий был впервые отделен от солей независимо двумя учёными: Ф. Вулером и А. А. Бази в 1828 году, в то время как литий был сепарирован Р. Бунзеном только в 1854 году, который, после изучения рубидия, отделил его спустя 9 лет. Цезий не был выделен в чистом виде вплоть до 1881 года, после того как Карл Сеттерберг подверг электролизу цианид цезия.
Твердость элементов, имеющих s-блок, в компактном виде (при обычных условиях) может варьироваться от очень малой (все щелочные металлы — их можно разрезать ножом) до довольно высокой (бериллий). Исключая бериллий и магний, металлы очень реакционноспособны и могут быть использованы в сплавах со свинцом в малых количествах (<2 %). Бериллий и магний, ввиду их высокой стоимости, могут быть ценными компонентами для деталей, где требуется твёрдость и лёгкость. Эти металлы являются чрезвычайно важными, поскольку позволяют сэкономить средства при добыче титана, циркония, тория и тантала из их минеральных форм; могут находить своё применение как восстановители в органической химии. Опасность и хранение
Все элементы, имеющие s-оболочку, являются опасными веществами. Они пожароопасны, требуют особого пожаротушения, исключая бериллий и магний. Храниться должны в инертной атмосфере аргона или углеводородов. Бурно реагируют с водой, продуктом реакции является водород, например:
,
![]()
исключая магний, который реагирует медленно, и бериллия, который реагирует только когда его оксидная плёнка снята с помощью ртути. Литий имеет схожие свойства с магнием, так как находится, относительно периодической таблицы, рядом с магнием. Электронная конфигурация — формула расположения электронов по различным электронным оболочкам атома химического элемента или молекулы.
С точки зрения квантовой механики электронная конфигурация — это полный перечень одноэлектронных волновых функций, из которых с достаточной степенью точности можно составить полную волновую функцию атома (в приближении самосогласованного поля).
Вообще говоря, атом, как составную систему, можно полностью описать только полной волновой функцией. Однако такое описание практически невозможно для атомов сложнее атома водорода — самого простого из всех атомов химических элементов. Удобное приближённое описание — метод самосогласованного поля. В этом методе вводится понятие о волновой функции каждого электрона. Волновая функция всей системы записывается как надлежащим образом симметризованое произведение одноэлектронных волновых функций. При вычислении волновой функции каждого электрона поле всех остальных электронов учитывается как внешний потенциал, зависящий в свою очередь от волновых функций этих остальных электронов.
В результате применения метода самосогласованного поля получается сложная система нелинейных интегродифференциальных уравнений, которая всё ещё сложна для решения. Однако уравнения самосогласованного поля имеют вращательную симметрию исходной задачи (то есть они сферически симметричны). Это позволяет полностью классифицировать одноэлектронные волновые функции, из которых составляется полная волновая функция атома.
Для начала, как в любом центрально симметричном потенциале, волновую функцию в самосогласованном поле можно охарактеризовать квантовым числом полного углового момента l и квантовым числом проекции углового момента на какую-нибудь ось m. Волновые функции с разными значениями m соответствуют одному и тому же уровню энергии, т. е. вырождены. Также одному уровню энергии соответствуют состояния с разной проекцией спина электрона на какую-либо ось. Всего для данного уровня энергии 2(2l + 1) волновых функций. Далее, при данном значении углового момента можно перенумеровать уровни энергии. По аналогии с атомом водорода принято нумеровать уровни энергии для данного l начиная с n = l + 1. Полный перечень квантовых чисел одноэлектронных волновых функций из которых можно составить волновую функцию атома и называется электронной конфигурацией. Поскольку всё вырожденно по квантовому числу m и по спину, достаточно только указывать полное количество электронов, находящихся в состоянии с данными n, l.
Расшифровка электронной конфигурации
По историческим причинам в формуле электронной конфигурации квантовое число l записывается латинской буквой. Состояние с l = 0 обозначается буквой s, l = 1 — p, l = 2 — d, l = 3 — f, l = 4 — g и далее по алфавиту. Слева от числа l пишут число n, а сверху от числа l — число электронов в состоянии с данным n, l. Например 2s2 соответствует двум электронам в состоянии с n = 2, l = 0. Из-за практического удобства (см. правило Клечковского) в полной формуле электронной конфигурации термы пишут в порядке возрастания квантового числа n, а затем квантового числа l, например 1s22s22p63s23p2. Поскольку такая запись несколько избыточна, иногда формулу сокращают до 1s22s2p63s2p2, т. е. опускают число n там, где его можно угадать из правила упорядочения термов.
Периодический закон и строение атома
Все занимавшиеся вопросами строения атома в любых своих исследованиях исходят из инструментов, которые предоставлены им периодическим законом, открытым химиком Д. И. Менделеевым; только в своём понимании этого закона физики и математики пользуются для истолкования зависимостей, показанных им, своим «языком» (правда, известен довольно ироничный афоризм Дж. У. Гиббса на этот счёт [1]), но, в то же время, изолированно от изучающих вещество химиков, при всём совершенстве, преимуществах и универсальности своих аппаратов ни физики ни математики, конечно, строить свои исследования не могут.
Взаимодействие представителей этих дисциплин наблюдается и в дальнейшем развитии темы. Открытие вторичной периодичности Е. В. Бироном (1915), дало ещё один аспект в понимании вопросов, связанных с закономерностями строения электронных оболочек. C. А. Щукарев, ученик Е. В. Бирона и М. С. Вревского, одним из первых ещё в начале 1920-х годов высказал мысль о том, что «периодичность есть свойство, заложенное в самом ядре».
При том, что полной ясности в понимании причин вторичной периодичности нет до сих пор, существует взгляд на эту проблему, подразумевающий то, что одной из важнейших причин этого феномена является открытая С. А. Щукаревым кайносимметрия — первое проявление орбиталей новой симметрии (др.-греч. кбйньт — новый и др.-греч. ухммефсЯб — симметрия; «кайносимметрия», то есть «новая симметрия»). Кайносимметрики — водород и гелий, у которых наблюдается орбиталь s, — элементы от бора до неона (орбиталь — р), — элементы первого переходного ряда от скандия до цинка (орбиталь — d), а также — лантаноиды (термин предложен С. А. Щукаревым, как и актиноиды) (орбиталь — f). Как известно, элементы, являющиеся кайносимметриками, во многих отношениях имеют физико-химические свойства, отличные от свойств других элементов, принадлежащих к той же самой подгруппе.
Ядерная физика дала возможность снять противоречие, связанное с «запретом» Людвига Прандтля [2].. В 1920-е же годы С. А. Щукарев сформулировал правило изобарной статистики, которое гласит, что в природе не может быть двух стабильных изотопов с одинаковыми массовым числом и зарядом атомного ядра, отличающихся на единицу — один из них обязательно радиоактивен. Законченную форму эта закономерность приобрела в 1934 году благодаря австрийскому физику И. Маттауху, и получила имя правила запрета Маттауха-Щукарева.[3][4]