
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Реферат: Энергия Гиббса
Реферат: Энергия Гиббса
ПЛАН
ВВЕДЕНИЕ ................................................................................................ 2
ЭНЕРГИЯ ГИББСА .................................................................................... 3
ЗАКЛЮЧЕНИЕ ........................................................................................ 14
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ................................... 15
ВВЕДЕНИЕ
В своем реферате я расскажу об энергии Гиббса.
 Гиббс Джозайя Уиллард (1839-1903), американский физик-теоретик, один из
создателей термодинамики и статистической механики. Разработал теорию
термодинамических потенциалов, открыл общее условие равновесия гетерогенных
систем — правило фаз, вывел уравнения Гиббса — Гельмгольца, Гиббса — Дюгема,
адсорбционное уравнение Гиббса. Установил фундаментальный закон статистической
физики — распределение Гиббса. Предложил графическое изображение состояния
трехкомпонентной системы (треугольник Гиббса). Заложил основы термодинамики
поверхностных явлений и электрохимических процессов. Ввел понятие адсорбции.
Гиббс Джозайя Уиллард (1839-1903), американский физик-теоретик, один из
создателей термодинамики и статистической механики. Разработал теорию
термодинамических потенциалов, открыл общее условие равновесия гетерогенных
систем — правило фаз, вывел уравнения Гиббса — Гельмгольца, Гиббса — Дюгема,
адсорбционное уравнение Гиббса. Установил фундаментальный закон статистической
физики — распределение Гиббса. Предложил графическое изображение состояния
трехкомпонентной системы (треугольник Гиббса). Заложил основы термодинамики
поверхностных явлений и электрохимических процессов. Ввел понятие адсорбции.
ЭНЕРГИЯ ГИББСА
В начале своей работы я думаю необходимо представить основные понятия теории Гиббса.
ПРАВИЛО ФАЗ ГИББСА в термодинамике: число равновесно сосуществующих в какой-либо системе фаз не может быть больше числа образующих эти фазы компонентов плюс, как правило, 2. Установлено Дж. У. Гиббсом в 1873-76.
ГИББСА ЭНЕРГИЯ (изобарно-изотермический потенциал, свободная энтальпия), один из потенциалов термодинамических системы. Обозначается G, определяется разностью между энтальпией H и произведением энтропии S на термодинамическую температуру Т: G = H — T·S. Изотермический равновесный процесс без затраты внешних сил может протекать самопроизвольно только в направлении убывания энергии Гиббса до достижения ее минимума, которому отвечает термодинамическое равновесное состояние системы. Названа по имени Дж. У. Гиббса.
ПОТЕНЦИАЛЫ ТЕРМОДИНАМИЧЕСКИЕ, функции объема, давления, температуры, энтропии, числа частиц и других независимых макроскопических параметров, характеризующих состояние термодинамической системы. К потенциалам термодинамическим относятся внутренняя энергия, энтальпия, изохорно-изотермический потенциал (Гельмгольца энергия), изобарно-изотермический потенциал (Гиббса энергия). Зная какие-либо потенциалы термодинамические как функцию полного набора параметров, можно вычислить любые макроскопические характеристики системы и рассчитать происходящие в ней процессы.
РАСПРЕДЕЛЕНИЕ ГИББСА каноническое, распределение вероятностей различных состояний макроскопической системы с постоянным объемом и постоянным числом частиц, находящейся в равновесии с окружающей средой заданной температуры; если система может обмениваться частицами со средой, то распределение Гиббса называется большим каноническим. Для изолированной системы справедливо Гиббса распределение микроканоническое, согласно которому все микросостояния системы с данной энергией равновероятны. Названо по имени открывшего это распределение Дж. У. Гиббса.
Реакции присоединения радикалов к непредельным соединениям лежат в основе современной технологии получения полимеров, сополимеров и олигомеров. Эти реакции протекают при крекинге углеводородов, галоидировании олефинов, окислении непредельных соединений. Они широко используются в синтезе разнообразных соединений и лекарственных препаратов. Реакции присоединения атомов водорода и гидроксильных соединений к непредельным и ароматическим соединениям сопровождают фотолиз и радиолиз органических материалов и биологических объектов.
|
В реакции радикального присоединения типа |
® |
XCH2C·HY |
рвется двойная С=С-связь и образуется
связь С- X. Как правило, образующаяся
s -связь прочнее рвущейся p -С- С-связи, и поэтому реакция присоединения экзотермична.
Это четко видно из сравнения энтальпии реакции DН и прочности образующейся связи D (Et-X) в табл. 1.
Другой важный фактор, влияющий на энтальпию реакции, -энергия стабилизации образующегося радикала XCH2C·H2Y: чем больше эта энергия, тем больше теплота присоединения радикала X· к олефину. Энергию стабилизации можно охарактеризовать, например, разницей прочности связей C- H в соединениях Pr- H и EtYHC- H. Ниже приведены данные, характеризующие вклад энергии стабилизации радикала CH3CH2C· H2Y, образующегося в результате присоединения метильного радикала к мономеру CH2=CHY, в энтальпию этой реакции.
Таблица 1.
Энтальпия, энтропия и энергия Гиббса присоединения атомов и радикалов X·к этилену.
|
X· |
-DH, кДж моль-1 |
-DS, Дж моль-1 К-1 |
-DG (298 K), кДж моль-1 |
|
H· |
150 | 84 | 125 |
|
Cl· |
82 | 88 | 56 |
|
C· H3 |
100 | 122 | 64 |
|
Me2C· H |
92 | 134 | 52 |
|
PhC· H2 |
63 | 122 | 27 |
|
N· H2 |
81 | 109 | 49 |
|
HO· |
122 | 100 | 93 |
|
CH3O· |
82 | 118 | 46 |
|
HO2· |
63 | 134 | 23 |
| Y | H | C(O)OMe | Cl | CN | Ph |
|
DPr- H - |
0.0 | 23.2 | 24.1 | 33.6 | 57.9 |
|
-DН,кДж моль-1 |
95.8 | 102.0 | 104.3 | 129.7 | 143.0 |
Видно, что чем больше энергия стабилизации радикала, тем меньше энтальпия реакции.
Все реакции присоединения протекают с уменьшением энтропии, т. к. происходит соединение двух частиц в одну (см. табл. 8.1). В силу этого для реакций присоединения энергия Гиббса, и при достаточной высокой температуре экзотермическая реакция присоединения является обратимой, т. к. DG = DH-TD S.
На любой процесс (реакцию) действуют два фактора:
Энатльпийный (экзо- или ендо) – Δ H;
Энтропильный (ТΔS).
При объединении этих двух факторов получаем:
ΔН – ТΔS = ΔG
G = H – TS – Энергия Гиббса.
Физический смысл Энергии Гиббса:
Ø если изменения ΔGр,т меньше нуля – то самопроизвольно идет процесс в заданном направлении;
Ø если изменения ΔGр,т больше нуля – самопроизвольно идет обратный процесс, а прямая реакция не идет совсем;
Ø
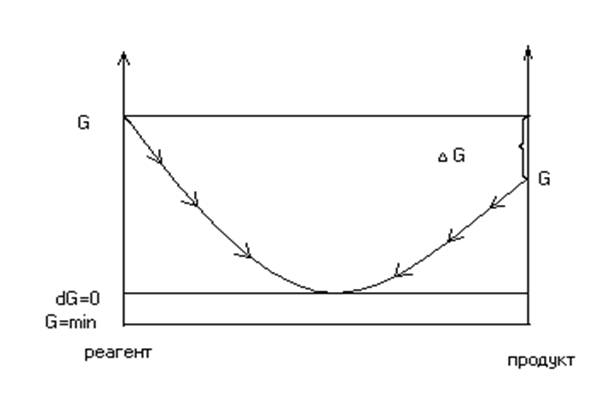 |
если изменения ΔGр,т равна нулю – это важнейшее термодинамическое равновесие.
Вывод: состояние термодинамического равновесия чрезвычайно устойчиво, так как при постоянстве Р, Т система выйти из равновесного состояния не может, так как выход равен возрастанию энергии Гиббса.
Чтобы система вышла из состояния равновесия необходимо изменить какие-либо внешние факторы (Р, Т, концентрация и так далее).
Есть понятие стандартное состояние Гиббса:
ΔGf0 298 [кДж / моль] – справочная величина.
Пользуясь справочными данными можно рассчитать изменение энергии Гиббса любого процесс.
ΔG 298 = ΣniΔ * ΔGf0 298 – ΣnjΔ * ΔGf0 298
продукт реагент
большинство процессов протекает при t более высоких чем стандартная (298). Для пересчета энергии Гиббса на более высокие температуры необходимы справочные данные по теплоемкостям, данные представленные в виде зависимости от температуры.
В справочниках эти данные обычно представлены в виде степенного ряда.
Cp0 = a + bT + cT2 + c’Т-2
где a, b, c, c’ – для каждого вещества свои.
Когда необходимо рассчитать для процесса
ΔCp0 = Δa + ΔbT + ΔcT2 + Δc’Т-2
Где Δa, Δb, Δc, Δc’ - будучи функциями состояния, рассчитываются по формулам:
Δa = Σniа - Σnjа
продукт реагент
Δb = Σnib - Σnjb
продукт реагент
Δc = Σnic - Σnjc
продукт реагент
Термодинамика фазовых равновесий. Фазовые равновесия в гетерогенных системах. Правило фаз Гиббса.
К фазовым равновесиям относятся переходы типа:
Ø Твердая фаза в равновесии с жидкостью (плавление – кристаллизация);
Ø Жидкая фаза в равновесии с паром (испарение – конденсация);
Ø Твердая фаза в равновесии с паром (возгонка – сублимация).
Основные понятия правила фаз:
Фаза (Ф) – это часть системы, имеющая границы раздела с другими ее частями.
Компонент (к) – это химически однородная составляющая системы, обладающая всеми ее свойствами.
Число степеней свободы (С) – это число независимых переменных которые можно произвольно менять не меняя числа фаз в системе.
(С, Ф, К) С = К – Ф +2
Существует правило фаз Гиббса.
Различают однокомпонентные, двухкомпонентные, трехкомпонентные системы (К=1, К=2, К=3).
К=1.
Сmin = 1 – 3 + 2 = 0
Cmax = 1 – 1 + 2 = 2
Для описания однокомпонентных систем выбрали координаты:
![]() Р (давление насыщенного пара)
Р (давление насыщенного пара)
![]() Т
(температура)
Т
(температура)
dP / dT = ΔHф.п. / (Tф.п. * ΔV)
эта зависимость сохраняется в силе для абсолютно всех фазовых переходов.


 Р
c
Р
c
![]()
![]() Тв. Ж. a
Тв. Ж. a
![]()
![]()
![]()

![]() b Пар
b Пар
![]()
Т кр. Т
Каждая линия диаграммы отвечает своему фазовому переходу:
Оb Тв. – Ж.
Оа Ж. - Пар
Ос Тв. - Пар
Поля диаграммы: твердая фаза, жидкая фаза, пар.
Т кр.: Пар – Газ
Поле фазы:
С = 2 (на полях Сmax)
C = 1 (на линиях)
Точка О – отвечает равновесию трех фаз: Тв. – Ж – Пар.
С = 0 – это значит, что нельзя менять ни температуру ни давление.
Остановимся теперь на химическом потенциале — величине, определяющей термодинамические характеристики не системы в целом, а одной молекулы в этой системе.
Если добавлять в систему молекулу за молекулой при постоянном давлении, то на добавление каждой новой частицы надо затратить в точности ту же работу, что на добавление любой предыдущей: объем системы будет расти, а плотность системы — и интенсивность взаимодействий в ней — меняться не будет. Поэтому термодинамическое состояние молекулы в системе удобно определять величиной свободной энергии Гиббса G, деленной на число молекул N,
|
m = G/N |
называемой химическим потенциалом (а так как в жидкой или твердой фазе и невысоких давлениях F » G , то здесь m » F/N ). Если N означает не число молекул, а, как обычно, число молей молекул, то и m относится не к одной молекуле, а к молю молекул.
Химический потенциал — или, что то же самое, свободная энергия Гиббса в расчете на одну молекулу — нам пригодится во второй части сегодняшней лекции, когда речь пойдет о распределении молекул между фазами. Дело в том, что молекулы перетекают из той фазы, где их химический потенциал выше, в ту, где их химический потенциал ниже, — это понижает общую свободную энергию системы и приближает ее к равновесию. А в равновесии химический потенциал молекул в одной фазе равен химическому потенциалу тех же молекул в другой фазе.
В последнее время при изучении свойств пластифицированных систем были обнаружены экспериментальные факты, противоречащие общепринятым представлениям и в ряде случаев не получившие должного объяснения. Это касается термодинамики пластифицированных систем, определения температуры стеклования (Тс) и оценки свойств систем, содержащих относительно небольшие количества пластификатора. Факты эти имеют большое значение для практики и теории, они связаны с метастабильностью пластифицированных систем и с неправильным использованием некоторых методов изучения их свойств.
Известно, что все системы делятся на устойчивые или стабильные, неустойчивые или лабильные и метастабильные, которые наиболее распространены. Поэтому изучение теплофизических свойств метастабильных систем имеет большое значение.
Метастабильная система устойчива по отношению ко всем системам, бесконечно мало отличающимся от нее, но имеется по крайней мере одна система, по отношению к которой она неустойчива. Состояние А, обладающее наименьшей энергией Гиббса, является истинно устойчивым, а состояние Б, обладающее большей энергией Гиббса, - метастабильным состоянием по отношению к состоянию А. Однако для перехода системы из состояния Б в состояние А требуется преодолеть потенциальный барьер. Если энергия возмущения меньше потенциального барьера, то система остается в состоянии Б.
Стабильность таких систем
зависит от соотношения времени релаксации (р) и времени опыта (оп);
под временем опыта подразумевается не только время лабораторного опыта, но и
время хранения и эксплуатации изделия. Если
р >> оп, то система может находиться в метастабильном
состоянии неограниченное время и она ничем не отличается от истинно устойчивой
системы. Поэтому к ней не следует применять термин "неравновесная".
Наоборот, в настоящее время широко распространен термин "метастабильное
равновесие". Система в состоянии А находится в истинном равновесии, а
система в состоянии Б - в метастабильном равновесии.
Метастабильное состояние является типичным для полимерных систем вследствие
очень большого размера макромолекул полимеров и значительных р.
Такие системы, например, можно получить закалкой, т.е. быстрым охлаждением
полимера или полимерной смеси до температуры значительно ниже их Тс.
При этом не изменяется структура системы и сохраняется приданная ей при более
высокой температуре структура. Это означает, что система "помнит"
свое прошлое. Такие системы называют системами с "памятью".
Исследованию их свойств посвящено много работ, разрабатывается термодинамика
этих систем. Эти свойства зависят от предыстории систем. К системам с памятью
относятся все полимеры и полимерные композиции, находящиеся при температуре
намного ниже их Тс. Время релаксации происходящих в них процессов очень велико,
в связи с чем стеклообразные полимеры при Т << Тс рассматривают как равновесные.
К таким системам применимы законы классической термодинамики.
Большое значение имеет термодинамическое сродство полимера к пластификатору, которое оценивают теми же параметрами, что и сродство полимера к растворителям: величиной и знаком энергии Гиббса (G) смешения параметром взаимодействия Флори-Хаггинса (1), вторым вириальным коэффициентом (А2). Величину G можно определить двумя способами. Первый способ состоит в прямом определении G на основании экспериментальных данных по давлению пара пластификатора над пластифицированной системой или по давлению набухания. Пластификаторы являются труднолетучими жидкостями, поэтому измерение их малых давлений требует специальных методов. Метод эффузии, который для этой цели применяется, имеет много недостатков. Более точным является метод определения давления набухания, давно используемый при изучении свойств пластифицированных эфиров целлюлозы. Он был успешно применен при исследовании сродства вулканизаторов каучуков к различным растворителям.
Определение G пластифицированных полимеров можно осуществлять с помощью метода, предложенного для смесей полимеров. Для этого следует измерить G смешения полимера, пластификатора и их смесей с какой-либо низкомолекулярной жидкостью, неограниченно смешивающейся с ними. Энергию Гиббса смешения можно определять на основании данных по светорассеянию растворов. Этот метод, представленный Вуксом для системы жидкость- жидкость, впервые был использован для систем полимер-растворитель в работе.
Второй способ определения величины G состоит в расчете этого параметра на основании экспериментально измеренных энтальпии и энтропии смешения полимера с пластификатором. Ее рассчитывают по уравнению: G = H - TS. Энтальпию смешения рассчитывают по закону Гесса, как описано выше, энтропию смешения определяют на основании температурной зависимости теплоемкости пластифицированных систем, измеренной с помощью сканирующего калориметра. Этот метод заслуживает внимания. Однако в рамках классической термодинамики абсолютные значения энтропии можно получить только при экстраполяции экспериментальной температурной зависимости теплоемкости к абсолютному нулю. В работе это было сделано, а в работе использован приближенный способ расчета величин S0, когда все величины энтропии приняты без нулевых слагаемых. Это может привести к ошибкам. Из изложенного следует, что необходимо развивать различные методы, которые должны давать одинаковые результаты. Для этого необходимо результаты, полученные разными методами, сопоставлять и систематически обсуждать.
ЗАКЛЮЧЕНИЕ
В своей работе я рассмотрела энергию Гиббса и относящиеся к этой теории понятия. Я рассказала о термодинамические потенциалы, правила фаз, распределение Гиббса, энтальпию, энтропию и конечно саму энергию Гиббса.
Вклад Джозайи Уилларда Гиббса в науку имеет большое значение. Его труды и исследования послужили основой для научных разработок его последователей, а так же имеют практическое значение.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ:
1. Теплофизика метастабильных жидкостей. Свердловск, УНЦ АН СССР, 1987.
2. Пригожин И., Дефей Р. Химическая термодинамика. Пер. с англ. Под ред. В.А. Михайлова. Новосибирск, Наука, 1966.
3. Кубо Р. Термодинамика. Пер. с англ. Под ред. Д.М. Зубарева, Н.М. Плакиды. М. Мир, 1970.
4. Тагер А.А. Высокомолекул. соед., 1988, т. А30, № 7, с. 1347.
5. Тагер А.А. Физикохимия полимеров. М., Химия, 1978.
6. Новикова Л.В. и др. Пласт. массы, 1983, № 8, с. 60.