
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Курсовая работа: Синтез бензальацетона
Курсовая работа: Синтез бензальацетона
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ БЕНЗАЛЬАЦЕТОНА”
Курсовая работа
Выполнил: студент
_________________
(подпись)
Руководитель:
_________________
(подпись)
Работа защищена
“___“ __________ 2007г.
Оценка _______
Зав. кафедрой: доцент, д. х. н.
_________________
(подпись)
Самара, 2007 г.
Содержание
1. Введение....................................................................................................... 3
1.1. Свойства бензальацетона................................................................... 3
1.2. Применение бензальацетона.............................................................. 3
1.3. Методика синтеза ............................................................................... 4
2. Литературный обзор................................................................................. 5
2.2. Способы получения……..……………………………………..…………...7
2.3. Химические свойства................................................................................ 9
Выводы.......................................................................................................... 17
Список литературы....................................................................................... 18
1. Введение
1.1. Свойства бензальацетона[14]
Бензальацетон (метилстирилкетон) — бесцветное или светло-желтое кристаллическое вещество в виде пластинок; по запаху напоминает кумарин; растворим в спирте, эфире, хлороформе, бензоле. Вызывает раздражение кожи. Температура плавления 42°С; молекулярная масса 148; температура кипения 260°С; 126 °С (9 мм. рт. ст.); d1515—1,0377; пD15—1,5836.
УФ спектр (в этаноле) λмакс lg(ε): 222нм (4,08), 284 нм (4,37).
Обладает характерными для α,β-непредельных кетонов химическими свойствами.
1.2. Применение бензальацетона
Применяется в парфюмерии в незначительных количествах, главным образом для духов. Является промежуточным продуктом для получения коричной кислоты. Используется в органическом синтезе. Бензальацетон производился в СССР с 1929 года. Количественное определение бензальацетона ведется по способу Мессенгера, описанному для количественного определения ацетона[15].
1.3. Методика синтеза [14]
Реактивы. Бензальдегид свежеперегнанньй 10 мл; ацетон 20 мл; гидроксид натрия (10%-ный раствор); соляная кислота; бензол.
При работе с бензальацетоном необходимо соблюдать осторожность, он сильно раздражает кожу!
В стакане емкостью 100 мл, снабженном мешалкой и термометром, смешивают 20 мл ацетона (значительный избыток ацетона необходим для предотвращения образования дибензальацетона), 10 мл свежеперегнанного бензальдегида и 10 мл воды. Стакан с реакционной смесью помещают в водяную баню и, поддерживая температуру смеси 25—30°С, приливают в нее при перемешивании постепенно 2,5 мл 10%-ного раствора гидроксида натрия. Смесь перемешивают 2 ч при комнатной температуре. После окончания реакции конденсации к реакционной массе приливают разбавленную соляную кислоту до кислой реакции на лакмус и смесь переливают в делительную воронку. Верхний слой — органический — в виде желтого масла отделяют от водного слоя. Водный слой переливают в маленькую делительную воронку и экстрагируют 8 мл бензола; бензольную вытяжку соединяют с органическим слоем из смеси, объединенный органический раствор в делительной воронке промывают 5 мл воды и отделяют водный слой. Сначала отгоняют бензол на водяной бане, потом бензальацетон в вакууме. Причем первые несколько миллилитров дистиллята, имеющего зеленоватый оттенок, отделяют. Собирают фракцию с температурой кипения 148—160°С (25 мм рт. ст.), имеющую светло-желтый оттенок. Бензальацетон при стоянии кристаллизуется. Выход бензальацетона 10 г.
2. Литературный обзор.
2.1. Способы получения
Получение альдегидов и кетонов
Поскольку как альдегиды, так и кетоны содержат карбонильную группу, то многие методы их получения сходны. Наряду с этим существует ряд особых способов, пригодных для синтеза только альдегидов или только кетонов[4].
1. Окисление или дегидрирование первичных или вторичных спиртов
Важное значение имеет метод окисления по Оппенауэру (1937 г.). Он представляет собой процесс, обратный реакции восстановления по Мейервейну — Пондорфу — Верлею, и применяется в основном для получения кетонов из вторичных спиртов. Последние вводят в реакцию с ацетоном в присутствии изопропилата или трет-бутилата алюминия.
Вначале образуется алкоголят алюминия вторичного спирта, дающий комплекс с ацетоном. Внутри последнего происходит перенос гидрид-иона от алкоксидного остатка на карбонильную группу.
Первичные спирты окисляются в альдегиды в том случае, если вместо ацетона в качестве акцептора гидрид-иона используют 1,4-бензохинон (п-хинон). В условиях реакции Оппенауэра двойные связи С=С не затрагиваются, что позволяет получать непредельные альдегиды и кетоны.
2. Оксосинтез (гидроформилирование, Ройлен, 1938 г.).
При температурах от 30 до 200 °С и давлении 100—400 кгс/см2 (1*1О7 до 4*107 Па) в присутствии дикобальтоктакарбонила олефины присоединяют водород и монооксид углерода с образованием альдегидов. Обычно получается смесь изомеров.
В определенных условиях могут получаться также и кетоны.
3. Гидратация ацетиленов.
Алкины и циклоалкины гидратируются в разбавленных растворах серной кислоты в присутствии сульфата ртути (II). Присоединение протекает по правилу Марковникова и приводит всегда к получению кетонов, за исключением ацетилена, который дает ацетальдегид. На первой стадии тройная связь (возможно, скомплексированная) подвергается нуклеофилыюй атаке водой с промежуточным образованием винилового спирта, который таутомерно превращается в карбонильное соединение. В данном случае кето-енолъное таутомерное равновесие практически полностью сдвинуто в сторону образования альдегида или соответственно кетона.
До последнего времени гидратация ацетилена являлась важнейшим способом производства ацетальдегида.
4. Пиролиз солей карбоновых кислот (Пириа, 1856 г.).
При нагревании кальциевых, бариевых или ториевых солей карбоновых кислот до ~300°С образуются кетоны.
Смеси таких солей с формиатами (солями муравьиной кислоты) дают альдегиды.
В промышленности исходят из самих карбоновых кислот, пары которых при 300 °С пропускают над оксидным марганцевым (II) (для альдегидов) или оксидными кадмиевыми(II) или ториевыми(IV) катализаторами (для кетонов).
5. Восстановление по Розенмунду (1918 г.).
Получение альдегидов прямым восстановлением карбоновых кислот затруднительно, так как обычно процесс не останавливается на стадии образования альдегида и идет дальше. По Розенмунду альдегиды получают гидрированием ацихлоридов над палладием, нанесенным на сульфат бария. В целом ряде случаев катализатор дезактивируют добавками хинолина с серой или тиомочевиной для предотвращения восстановления двойной связи С=О.
6. Восстановление по Стефену (1925 г.)
При восстановлении нитрилов хлоридом олова (II) в соляной кислоте также получаются альдегиды. Промежуточно при этом образуются альдимины, частично выкристаллизовывающиеся в виде гексахлорстаннатов(IV).
В качестве восстановителя все чаще используют также алюмогидрид лития. Это соединение, однако, не следует брать в избытке, поскольку тогда альдимин будет восстанавливаться далее до амина
7. Взаимодействие сложных эфиров с реактивом Гриньяра.
Реакция эфиров муравьиной кислоты с алкил- или арилмагнийгалогенидами дает альдегиды.
Следует избегать избытка реактива Гриньяра; в противном случае образуются вторичные спирты. Поэтому на практике раствор магнийор-ганического соединения прибавляют по каплям к эфиру муравьиной кислоты. Для этих же целей можно использовать и ортоэфиры муравьиной кислоты; в таком случае получаются ацетали, которые далее гидролизуются до альдегидов.
Из эфиров других кислот, в частности и ортоэфиров, получаются кетоны, в последнем случае промежуточно образуются кетали.
Синильная кислота или нитрилы при взаимодействии с реактивом Гриньяра также дают альдегиды или соответственно кетоны.
8. Взаимодействие ацилхлоридов с алкилкадмиевыми соединениями
Диалкилкадмиевые производные используют в синтезе кетонов из ацилгалогенидов, так как в отличие от магнийорганических соединений они не реагируют с карбонильными группами. При синтезах их можно генерировать непосредственно в реакционной смеси.
9. Реакция Сомле (1913 г.).
Хлорметиларены реагируют в водноэтанольном растворе с уротропином с образованием альдегидов.
10. Реакция Этара (1877 г.)
При обработке алкиларенов хромилхлоридом в четыреххлористом углероде образуются альдегиды или кетоны. Например, толуол окисляется до бензальдегида.
Метилареиы можно также окислять действием оксида хрома (VI) в уксусном ангидриде.
11. Ацилирование по Фриделю — Крафтсу (1877 г.)
При взаимодействии аренов с ацилгалогенидами в присутствии кислот Льюиса, таких как хлорид алюминия, получают кетоны.
Ацилирование может быть и внутримолекулярным. В качестве примера приведем синтез инданона-1.
Вместо ацилгалогенидов используются также сами кислоты или их ангидриды. В противоположность алкилированию по Фриделю — Крафтсу, при котором берутся лишь каталитические количества катализатора, при проведении ацилирования по Фриделю — Крафтсу необходимо брать более чем стехиометрические количества катализатора. Это связано с тем, что как ацилирующий агент, так и продукт реакции образуют с катализатором комплекс в соотношении 1:1.
Ацилирование аренов по Фриделю — Крафтсу относится к SЕ-реакциям. В зависимости от условий реагентом является либо комплекс состава 1:1, либо ацил-катион.
В аналогичных условиях ацилируются циклоалканы и олефины. В последнем случае наряду с продуктами ацилировакия получаются также продукты присоединения.
Родственными ацилированию по Фриделю — Крафтсу являются следующие реакции, при которых бензоидный атом водорода замещается на альдегидную группу (формилирование).
12. Реакция Гаттермана — Коха (1887 г.)
Арены формилируют смесью моноксида углерода и хлористого водорода в присутствии хлорида алюминия и хлорида меди(I). В противоположность существовавшим ранее представлениям этот синтез альдегидов протекает не через промежуточное образование нестабильного при комнатной температуре формилхлорида, а с участием ацилиевого комплекса, образующегося непосредственно из реагентов.
Роль хлорида меди(I) состоит в первоначальном образовании комплекса с моноксидом углерода, что повышает стационарную концентрацию последнего.
Фенолы и их простые эфиры, а также нитробензол и другие соединения с сильными электроноакцепторными заместителями не вступают в эту реакцию.
13. Реакция Гаттермана (1906 г.)
По этому способу формилирования арены вводят во взаимодействие с синильной кислотой и хлористым водородом. Катализатором служит хлорид алюминия. В реакцию Гаттермана вступают фенол и его простые эфиры, но не вступают нитробензол и ароматические амины.
Вполне возможно, что в данном случае формилирующим агентом является соединение:
![]()
Использования ядовитой синильной кислоты можно избежать, если по Адамсу (1923 г.) в реакцию вводить цианид цинка и хлористый водород.
14. Реакция Вильсмейера — Хаака (1927 г.)
Реакционноспособные и прежде всего многоядерные арены, а также фенолы и их простые эфиры формилируются с образованием альдегидов под действием N,N-дизамещенных формамидов, например N-метилформанилида или N,N-диметилформамида и оксихлорида фосфора.
Промежуточно образующийся комплекс формамида и оксихлорида фосфора в случае N-метилформанилида может быть выделен в свободном виде.
2.2. Химические свойства
1. Реакции с водой и спиртами.
При присоединении к альдегидам воды образуются гидраты (1,1-диолы).
В большинстве случаев это равновесие сильно сдвинуто влево, в сторону исходных реагентов, так что выделить геминальные диоксисоединения невозможно (правило Эрленмейера). Тем не менее такие реакции, как олигомеризация формальдегида и ацетальдегида, протекают с участием гидратов. Соединения, у которых по соседству с карбонильной группой находится электроноакцепторный заместитель, образуют устойчивые гидраты. Примерами таких соединений служат хлораль, трикетоиндан и аллоксан[4].
Присоединение к альдегидам спиртов приводит к полуацеталям, их образование катализируется кислотами и основаниями. В присутствии сильных кислот в результате дальнейшей реакции с новой молекулой спирта образуются ацетали.
Получить кетали из кетонов, присоединяя к ним спирты, нельзя, поскольку равновесие сильно сдвинуто влево. Однако простейшие кетали можно получить при взаимодействии кетонов с ортоэфирами муравьиной кислоты (Хельферих, 1924 г.).
В кислой среде ацетали и кетали гидролизуются до альдегидов и кетонов. Однако по отношению к основаниям они стабильны и используются для защиты карбонильной группы.
2. Реакция с тиоспиртами.
При взаимодействии альдегидов и кетонов с тиоспиртами (меркаптанами) можно получить меркаптали. С этандитиолом-1,2 образуются циклические меркаптали (1,3-дитиоланы).
В присутствии скелетного никеля 1,3-дитиоланы подвергаются гидрогенолизу. Таким путем карбонильные соединения можно восстановить до углеводородов.
3. Образование бисульфитных соединений.
При обработке концентрированным водным раствором бисульфита натрия альдегиды и кетоны образуют соли оксисульфокислот, так называемые бисульфитные соединения.
Бисульфитные соединения плохо растворимы и используются для отделения альдегидов и кетонов. Нагревание этих соединений с разбавленными кислотами или с водным раствором карбоната натрия приводит к регенерации карбонильных соединений.
4. Реакции с аминосоединениями.
Соединения структуры Z—NH2 легко присоединяются к альдегидам и кетонам. Однако в большинстве случаев продукты присоединения неустойчивы и легко дегидратируются через промежуточные карбений-иммониевые ионы.
При взаимодействии альдегидов с аммиаком, протекающем через стадию малостабильных альдегидаммиаков, образуются альдимины.
Альдимины обычно тримеризуются в гексагидро-1,3,5-триазины.
Реакция с формальдегидом протекает сложнее. Бензальдегид также реагирует иначе, взаимодействие его с аммиаком приводит к гидробензамиду.
В случае таких кетонов, как ацетон, аммиак вызывает альдольную реакцию. Образующиеся при этом димеры или тримеры далее реагируют с аммиаком с образованием диацетонамина (4-амино-4-метилпентанона-2) или триацетонамина (2,2,6,6-тетраметил-пиперидон-4).
Первичные амины конденсируются с альдегидами в азометины (основания Шиффа).
В большинстве случаев азометины представляют собой кристаллические соединения и используются для выделения, очистки и идентификации альдегидов.
Большее значение для этой цели имеют гидразоны, фенилгидразоны, 4-нитрофенилгидразоны и 2,4-динитрофенилгидразоны, образующиеся при взаимодействии альдегидов и кетонов с гидразином или соответствующим замещенным гидразином. При реакции с гидразином могут получаться также кристаллические азины.
При нагревании с гидроксидом калия в триэтиленгликоле до ~200°С или при действии трет-бутилата калия в диметилсульфоксиде гидразоны теряют азот и образуют углеводороды (реакция Вольфа—Кижнера, 1912 г.). Реакция, вероятно, протекает по следующему механизму:
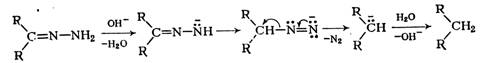
Гидроксиламин, а также семикарбазид конденсируются с альдегидами и кетонами.с образованием соответственно оксимов и семикарбазонов, также используемых для идентификации карбонильных соединений.
Азометины, гидразоны, азины, оксимы и семикарбазоны более или менее легко могут быть гидролизованы обратно до исходных карбонильных соединений. Регенерация кетонов из 2,4-динитрофенилгидразонов легче всего осуществлять нагреванием с гидратом толуол-4-сульфокислоты в хлороформе. Оксимы, кроме того, являются промежуточными продуктами в ряде синтезов. Например, (Z)-альдоксимы дегидратируются до нитрилов. В противоположность этому (E)-диастереомеры вследствие стереоэлектронных эффектов не вступают в реакцию; в условиях дегидратации эти соединения претерпевают перегруппировку Бекмана до N-замещенных формамидов.
В присутствии серной кислоты, хлорида фосфора(V), полифосфорной кислоты или других катализаторов кетоксимы также претерпевают перегруппировку Бекмана (1886 г.) с образованием N-замещенных амидов карбоновых кислот. На примере оксимов несимметричных кетонов установлено, что при этом имеет место анти-перегрупаировка: гидро-ксильная группа меняется местами с остатком, находящимся к ней в транс-положении.
При действии на альдегиды и кетоны вторичных аминов первоначально также протекает присоединение. Если в α-положении к карбонильной группе имеется протон, то нестабильное промежуточное соединение стабилизуется, отщепляя воду и образуя енамин.
Енамины являются очень реакционноспособными соединениями и используются во многих синтезах. Если, как в случае бензальдегида, в α-положении к карбонильной группе не имеется атома водорода и, таким образом, отщепление воды невозможно, то промежуточно образующийся карбений-иммониевый ион присоединяет вторую молекулу амина с образованием аминаля.
5. Циангидринный синтез
Присоединение синильной кислоты в присутствии основных катализаторов к альдегидам и некоторым кетонам приводит к циангидринам (α-оксинитрилам).
Под действием щелочей циангидрины расщепляются, при кислом гидролизе образуют α-оксикарбоновые кислоты.
6. Альдольная реакция
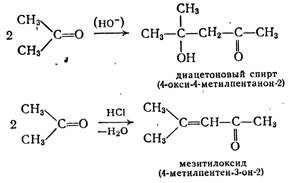
Взаимодействие альдегидов или кетонов (карбонильная компонента) самих с собой или с другими альдегидами и кетонами, выступающими в качестве С—Н-кислотной компоненты (метиленовой компоненты) с образованием (β-оксикарбонильных соединений, называют альдольной реакцией. Кислоты и основания катализируют эту реакцию. Например, в присутствии гидроксидов щелочных или щелочноземельных, металлов из ацетальдегида образуется ацетальдоль («аль» от альдегида и «ол» от спирта) 3-оксибутаналь.
Какая из стадий реакции (первая или вторая) будет определять скорость всего превращения, зависит от кинетической кислотности метиленовой компоненты и от электрофильности карбонильной компоненты. В случае альдолыюй реакции с ацетальдегидом наиболее медленной является первая стадия, в случае ацетона из-за меньшей реакционной способности его карбонильной группы — вторая стадия.
При повышенных температурах часто протекает дегидратация, приводящая к α,β-непредельным соединениям. Так, из ацетальдоля получают кротоновый альдегид.
При кислотном катализе альдольная реакция протекает через енольную форму, причем отщепление воды наблюдается почти всегда.
Примером альдолыюй реакции между различными альдегидами является образование пентаэритрита из ацетальдегида и формальдегида в присутствии гидроксида кальция. При этом в заключение протекает перекрестная реакция Канниццаро между образовавшимся альдегидом и молекулой формальдегида.
Аналогично альдегидам реагируют и кетоны, например:
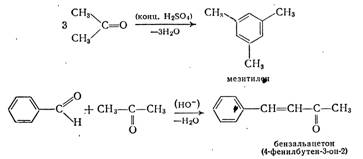
Как и в случае реакции бензальдегида с ацетоном, кетоны при реакции с альдегидами всегда играют роль метиленовой компоненты. В последнее время предпочитают использовать азометины альдегидов, поскольку для альдегидов, содержащих в α-положении атом водорода, предпочтительной является самоконденсация.
По схеме альдольной реакции протекают реакции и других С-Н-кислотных соединений с карбонильными соединениями. К ним относятся конденсация Кневенагеля, реакция Перкина и синтезы оксиранов (глицидный синтез) по Дарзану.
7. Бензоиновая конденсация
При действии водно-спиртовых растворов цианида калия арилальдегиды димеризуются в α-оксикетоны. Из бензальдегида образуется бензоин.
Считается доказанным следующий механизм этой реакции:
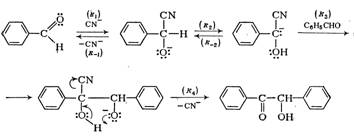
Как и в альдольной реакции, карбанион присоединяется к альдегиду.
Алифатические альдегиды дают в этих условиях не ацилоины, а альдоли, образования ацилоинов из этих соединений можно добиться, используя ферменты определенных видов дрожжей.
8. Реакция Канниццаро (1853 г.)
Альдегиды, не содержащие в α-положении к карбонильной группе атома водорода, т. е. преимущественно ароматические альдегиды, в присутствии гидроксидов щелочных или щелочноземельных металлов диспропорционируют до спирта и кислоты.
В случае альдегидов, имеющих α-атом водорода, гораздо быстрее протекает альдольная реакция. Формально атом водорода одной молекулы альдегида в виде гидрид-иона переносится к связи С=О другой молекулы. Начинается этот процесс с присоединения гидроксил-иона. Затем с участием атома металла образуется комплекс, внутри которого и происходит гидридный перенос.
Если в реакции принимают участие два различных альдегида, то говорят о перекрестной реакции Канниццаро. В таком случае формальдегид всегда играет роль донора гидрид-иона.
9. Реакция Кляйзена — Тищенкр (1906 г.)
При взаимодействии любых альдегидов с алкоголятом алюминия происходит гидридный перенос, поскольку основность этого реагента недостаточна для осуществления альдольной реакции. В результате образуются сложные эфиры карбоновых кислот.
Реакция также протекает через образование комплекса.
10. Восстановление по Мейервейну—Пондорфу—Верлею (1925, 1926 гг.)
В обращение окисления по Оппенауэру, при нагревании альдегидов и кетонов с каталитическими количествами изопропилата алюминия в растворе изопропанола происходит их восстановление, соответственно до первичных и вторичных спиртов. В образующемся комплексе происходит гидридный перенос от алкоксид-иона на карбонильное соединение. При этом устанавливается равновесие, сдвиг которого вправо осуществляют за счет непрерывной отгонки ацетона из реакционной смеси.
11. Аминометилирование (реакция Манниха, 1917 г.)
Кетоны, содержащие в α-положении атом водорода, как С-Н-кислоты аминометилируются при взаимодействии с формальдегидом (или другими альдегидами) и аммиаком или при взаимодействии с первичными или вторичными аминами. Реакция в большинстве случаев проводится в присутствии кислот, хотя возможен и основной катализ. Первоначально образуется карбений-иммониевый ион, реагирующий далее с енольной формой кетона.
Конечные продукты реакции Манниха называют основаниями Манниха.
12. Пинаконовое восстановление
При восстановлении кетонов натрием или амальгамами натрия или магния образуются 1,2-диолы (пинаконы, важнейшие спирты и фенолы, пинакон). К тем же результатам приводит и электрохимическое восстановление (катодное восстановление). Такого типа реакции идут на поверхности металлов и протекают через анион-радикалы, так называемые кетилы.
Бензальдегид при реакции с амальгамой натрия или цинка также может быть превращен в 1,2-диол.
13. Восстановление по Клёменсену (1913 г.)
При реакции с амальгамированным цинком и концентрированной соляной кислотой альдегиды и кетоны превращаются в углеводороды.
14. Окисление альдегидов
Альдегиды очень легко окисляются до карбоновых кислот, являясь таким образом восстановителями.
Так, они восстанавливают аммиачный раствор нитрата серебра (реактив Толленса) до серебра, а щелочной раствор гидроксида висмута (III) с добавкой комплексообразователя — винной кислоты (реактив Ньюленда) — до металлического висмута.
Алифатические альдегиды выделяют оксид меди(I) из щелочного раствора гидроксида меди(II), содержащего в качестве комплексообразователя тартрат калия-натрия (реактив Фелинга).
На воздухе бензальдегид подвергается автоокислению до бензойной кислоты. В данном случае протекает радикальный процесс, идущий через промежуточное образование среди других бензоильных радикалов и пербензойной кислоты.
15. Реакция Вильгеродта — Киндлера (1887, 1923 гг.)
При нагревании алкиларилкетонов в запаянных ампулах с водными растворами полисульфида аммония или, проще, с серой и первичными или вторичными аминами (чаще всего с морфолином) образуются амиды ω-арилкарбоновых кислот или сами кислоты. Механизм этой реакции очень сложен, промежуточно образуются енамины (Майер, 1964 г.).
Выводы
Таким образом, в данной работе рассмотрен бензальацетон, его свойства, способы получения и применение. Взаимодействием бензальдегида с ацетоном получен бензальацетон с выходом …г или …% масс. от теоретического. Определена температура плавления, показана сходимость полученных данных с литературными, а следовательно, очень низкая доля примесей в полученном бензальацетоне и чистота продукта синтеза.
Список литературы
1. Нейланд О.Я. Органическая химия, М.: Высшая школа, 1990, 751с.
2. Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
3. Физер Л., Физер М. Реагенты для органического синтеза. Том 2. М., Мир, 1970, 390.
4. З. Гауптман, Ю. Грефе, Х. Ремане Органическая химия, Москва, «Химия», 1979, 832
5. Кнунянц И.Л. Большая Российская энциклопедия, М.: 2003, 972с.
6. Юрьев Ю.К. Практические работы по органической химии. Выпуск 1 и 2. Изд. 3-е. М., Изд-во МГУ, 1964.
7. Юрьев Ю.К., Левина Р.Я., Шабаров Ю.С., Практические работы по органической химии. Выпуск 4. М. Из-во МГУ, 1969.
8. Шабаров Ю.С. Органическая химия: В 2-х кн. - М.:Химия, 1994.- 848 с.
9. Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. - 623 с.
10. Моррисон Р., Бойд. Органическая химия. - М.: Мир, 1974. - 1132 с.
11. Терней А. Современная органическая химия: В 2 т. - М.: Мир, 1981. - Т.1 - 670 с; Т.2 - 615 с.
12. Робертс Дж., Кассерио М. Основы органической химии: В 2 т. - 2-е изд. -М.: Мир, 1978. - Т.1 - 842 с; Т.2 - 888 с.
13. В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, - 708 с.
14. Птицина О.А., Куплетская Н.В., Тимофеева В.К. и др. Лабораторные работы по органическому синтезу. М.: Просвещение, 1979, 256с
15. Исагулянц В.И. Синтетические душистые вещества, 2 издание, Ереван, 1946, 831с.