
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Реферат: Группа пневмовирусов
Реферат: Группа пневмовирусов
Курсовая работа
на тему:
"Группа пневмовирусов"
Содержание
1. Уникальные особенности пневмовирусов
1.1 Общее введение
1.2 Номенклатура
1.3 Выделение вируса
1.4 Хранение вируса
2. Основные методы
2.1 Культивирование респираторно-синцитиального вируса
2.1.1 Чувствительные культуры клеток
2.1.2 Цитопатогенное действие
2.1.3 Методы повышения инфекционное ™
2.1.4 Методы стабилизации инфекционное
2.2 Методы определения инфекционности
2.3 Гемагглютинация
2.4 Гемадсорбция
2.5 Иммунофлуоресцентные методы
2.6 Твердофазный иммуноферментный анализ
3. Аналитические методы
3.1 Радиоактивное лечение, радиоиммуноанализ и радиоиммунопреципитация
3.1.1 Введение радиоактивной метки в вирусные белки в зараженных РСВ клетках
3.1.2 Радиоиммуноанализ
3.1.3 Радиоиммунопреципитация
3.2 Метод выявления дефектных интерферирующих частиц
3.3 Электронная микроскопия
3.3.1 Морфология вирионов
3.3.2 Морфогенез вирионов
3.3.3 Сканирующая электронная микроскопия
3.3.4 Локализация и количественное определение вирусных антигенов по связыванию стафилококков
3.4 Концентрирование вируса
3.5 Очистка и радиоактивное мечение РСВ
3.5.1 Градиентное центрифугирование
3.5.2 Очистка РСВ методом изопикнического центрифугирования
3.6 Радиоактивное мечение вирионной РНК
3.7 Анализ вирусных РНК и белков методом электрофореза в полиакриламидном геле
3.7.1 Анализ белков
3.7.2 Анализ РНК
3.8 Очистка матричной РНК для трансляции
3.9 Молекулярное клонирование и олигонуклеотидное секвенирование
3.10 Очистка вирусных белков
3.10.1 Вирусные полипептиды
3.10.2 Очистка капсидного белка
3.10.3 Очистка вирионных гликопротеинов
4. Специальные биологические методы
4.1 Получение моноклональных антител
4.2 Выделение температурно-чувствительных мутантов
4.3 Получение персистентно инфицированных клеточных культур
1. Уникальные особенности пневмовирусов
1.1 Общее введение
В состав группы пневмовирусов входят респираторно-синцитиальный вирус человека, респираторно-синцитиальный вирус крупного рогатого скота и вирус пневмонии мышей. Прототип этой группы, респираторно-синцитиальный вирус, - это плеоморфный оболочечный вирус, геном которого представлен несегментированной РНК негативной полярности. РСВ принадлежит роду Pneumovirus, который вместе с родами Paramyxovirus и Morbillivirus образует семейство Paramyxoviridae. Однозначно доказана этиологическая роль РСВ при ежегодных вспышках респираторных заболеваний у детей. Уникальность биологических, биохимических и клинических характеристик этого вируса не позволяет объединить его с представителями рода Paramyxovirus. РСВ обнаруживают у сельскохозяйственных животных; РСВ крупного рогатого скота вызывает у животных респираторное заболевание. Антитела к РСВ часто присутствуют и в сыворотке крови других домашних и лабораторных животных, контактирующих с человеком, но выделение вируса от этих животных не описано. Изоляты РСВ, выделенные от человека и крупного рогатого скота, имеют антигенное родство; среди человеческих изолятов не обнаружена связь между антигенной вариабельностью и клиникой заболеваний. Вирус пневмонии мышей, вызывающий у этих животных респираторное заболевание, не имеет антигенного родства с РСВ, однако в некоторой степени сходен с ним; во всяком случае это единственный вирус, достаточно близкий к РСВ, чтобы быть включенным в род Pneumovirus. Антитела к ВПМ с высокой частотой обнаруживаются в сыворотке крови человека; имеются неопубликованные данные, указывающие на то, что в лабораторные колонии мышей этот вирус попал от человека.
Основные свойства РСВ приведены в табл.1. РСВ отличается от других парамиксовирусов большим числом идентифицированных белков, порядком генов (имеются два гена неструктурных белков, один из которых расположен между З'-концевой лидерной последовательностью и геном нуклеопротеина, а другой - между гипотетическими генами гликопротеинов F и G), отсутствием гемагглютинина и нейраминидазы, а также характерными размерами нуклеокапсида и поверхностных выступов.
Таблица 1. Общие свойства пневмовирусов
|
Известные представители рода Нуклеиновая кислота Белки'> Вирионные ферменты Плавучая плотность частиц Морфология частиц Морфология нуклеокапсида Морфология мембранных выростов Генетические взаимодействия Стабильность Круг хозяев Патогенность Цитопатогенность |
РСВ человека РСВ крупного рогатого скота Вирус пневмонии мышей Однонитевая несегментированная РНК с коэффициентом седиментации 50S; мол. масса >5 - 10е 2 гликопротеина оболочки: гликопротеин G мол. масса 90 кД и связанные между собой дисульфидными связями продукты расщепления гликопротеина F 2 негликозилированных белка оболочки: белок матрикса мол. масса 26 кД и белок с неизвестной функцией мол. масса 24 кД 3 белка сердцевины: гипотетическая полимераза, нуклеокапсидный белок и фосфопротеин 3 неструктурных белка неизвестной функции РНК-полимеразная активность Гемагглютинин Нейраминидазная активность отсутствует 1.15-1,26 г/см3 Плеоморфные частицы диаметром 80^ - 500 нм и нитевидные частицы диаметром 60-ПО нм и длиной до 5 мкм. Спиральный тип симметрии, диаметр 12 - 13 нм Длина 10-12 нм 8 групп комплементации Рекомбинация или множественная реактивация не обнаружены Термолабильны, нестабильны при рН<3,0, чувствительны к замораживанию и оттаиванию In vivo, вероятно, ограничен Ассоциированы преимущественно с респираторными заболеваниями и спорадически с другими патологическими состояниями Образуют цитоплазматические, реже ядерные включения, часто формируют синцитии. Имеют склонность к персистенции |
1.2 Номенклатура
В англоязычной литературе для обозначения респираторно-синцитиального вируса имеет смысл использовать сокращение RS-вирус, а не RSV, чтобы отличать его от вируса саркомы Рауса, типичного представителя ретровирусов. Кроме того, респираторно-синцитиальный вирус крупного рогатого скота следует отличать от не родственного ему синцитий-образующего вируса, также относящегося к ретровирусам.
1.3 Выделение вируса
В связи с необычной лабильностью РСВ и низким титром в секретах и зараженных тканях при выделении этого вируса требуется особая тщательность. Попытки выделения вируса из хранившихся образцов редко бывают успешными. Имеет смысл доставлять чувствительную культуру клеток непосредственно к месту нахождения пациента или зараженного животного, а не транспортировать содержащие вирус материалы в лабораторию для заражения культуры.
Чаще всего для выделения РСВ используют линии клеток Нер-2 и HeLa, однако описано использование культуры почек макак-резусов, линий BS-C-I, MRC-5, WI-38 и некоторых других. Лучшим материалом для выделения РСВ служат смывы слизистой носа или носоглотки больных детей. Смывы можно перенести в пробирку с небольшим количеством буферного раствора или бульона. Содержимое пробирок перемешивают энергичным встряхиванием. Образцы следует держать в ледяной бане и как можно скорее использовать для заражения чувствительной культуры клеток. Во всяком случае, промежуток между сбором материала и заражением культуры не должен превышать нескольких часов. Из антибиотиков наиболее часто используют пенициллин ', стрептомицин, неомицин и микостатин.
Для успешного выделения РСВ важно избегать замораживания и оттаивания образцов, при их хранении поддерживать достаточно низкую температуру, как можно быстрее использовать материал для заражения чувствительной культуры и включать в состав культуральной среды ЭТС или БСА. Сыворотка взрослых домашних животных, обычно используемая для культивирования клеток, как правило, содержит достаточно высокий титр нейтрализующих РСВ антител; поэтому ее нельзя использовать при выделении этого вируса. Зараженную культуру инкубируют при 36-37 °С и наблюдают за ней не менее 3 нед, регулярно меняя среду. Для выявления ЦПД могут потребоваться дополнительные слепые пассажи. Для РСВ характерно ЦПД в виде образования синцития, но этот эффект наблюдается не всегда, и его проявление зависит от штамма вируса и типа зараженной культуры.
1.4 Хранение вируса
Пневмовирусы следует хранить в интервале температур от -70°С до - 198°С. При - 20°С наступает достаточно быстрая потеря инфекционное™, и в таких условиях вирус нельзя хранить более 3 мес. Лиофилизация или добавление глицерина позволяют увеличить срок хранения вируса при - 20°С. Быстрота падения инфекционности зависит от природы содержащего вирус материала; так, вирус, связанный с мембранами, более стабилен, чем свободный.
2. Основные методы
2.1 Культивирование респираторно-синцитиального вируса
2.1.1 Чувствительные культуры клеток
Имеется большой набор клеточных линий, чувствительных к РСВ. В диагностических лабораториях чаще всего используют линию гетероплоидных клеток человека Нер-2, поскольку ее легко поддерживать.
Тем не менее для выделения РСВ более подходят диплоидные линии клеток человека, например эмбриональных клеток легких WI-38, HeL-1, L-4, L-49, HLDC-1, - 4, - 6 и эмбриональных клеток мозга НВС-1 и НВС-4.
РСВ размножается и в разнообразных культурах клеток животных, в частности в клетках почек кошек, норок и кенгуровой крысы. Степень чувствительности разных линий клеток, по-видимому, зависит от схемы пассирования используемого штамма вируса. Например, после трех пассажей через, индивидуальные бляшки на культуре клеток WI-38 полученный изолят вируса давал на этих клетках в 50 раз более высокий титр, чем вирус, пассируемый на клетках BS-C-1.
РСВ может быть адаптирован и к росту в некоторых первичных культурах клеток холоднокровных животных, например черепахи Testudo graced, но он не размножается в клетках земноводных и беспозвоночных, в частности комаров. В отличие от многих других парамиксовирусов РСВ не размножается в куриных эмбрионах и культуре клеток куриного эмбриона. Кроме того, в отличие от вируса пневмонии мышей он не размножается в клетках ВНК-21.
При серийном пассировании РСВ с высокой частотой образуются дефектные интерферирующие частицы. Разработан простой колориметрический метод обнаружения и количественного определения ДИЧ,
Для размножения РСВ в культуре клеток можно использовать основную минимальную среду Игла, желательно с дополнительными аминокислотами. Лучший выход вируса, как правило, дают активно делящиеся клетки. Можно использовать и другие среды, например среду Лейбовича, для поддержания рН которой не требуется газообразная СОг. Температура размножения РСВ составляет 25-39 °С; максимальный выход получают при температуре около 37°С.
2.1.2 Цитопатогенное действие
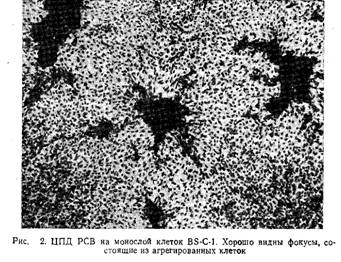
Характерное для РСВ цитопатогенное действие - это образование синцития. В синцитиях ядра клеток часто образуют агрегаты, окружающие центральную полость. Однако формирование синцития наблюдается далеко не всегда; характер ЦПД определяется многими факторами, в том числе штаммом вируса, условиями культивирования и типом клеток. Например, при заражении клеток почек африканской зеленой мартышки образуются фокусы агрегированных клеток. Под агаровым покрытием они выглядят как интенсивно окрашенные сгустки клеток, напоминающие фокусы трансформации, индуцируемые онкогенными вирусами. Однако индуцируемые РСВ фокусы окружены слабо окрашенной областью (рис.2). Ее образование обусловлено миграцией клеток к формирующемуся скоплению. Фокусы состоят из увеличившихся в размерах разбухших клеток, которые затем дают начало синцитию. Несмотря на внешнее сходство фокусов, индуцируемых онкогенными вирусами и РСВ, клетки последних не сохраняют способность к делению.
2.1.3 Методы повышения инфекционное ™
Обработка вируса или клеток ДЭАЭ-декстраном повышает чувствительность клеток к РСВ. При оптимальной концентрации, равной 40 мкг/мл, ДЭАЭ-декстран в 2 раза повышает выход вируса; имеются данные и о том, что число клеток, зараженных РСВ крупного рогатого скота, возрастает в этих условиях в 11 раз. Нисевич и др. сообщили о 100-кратном увеличении числа клеток, зараженных РСВ человека, в присутствии 15 мкг/мл ДЭАЭ-декстрана.
2.1.4 Методы стабилизации инфекционное
Инфекционность РСВ стабилизируется в присутствии высоких концентраций сахарозы или ионов магния. Ферни и Герин сообщили, что в присутствии 1 М MgS04 стабильность РСВ возросла в 30 раз и период полужизни инфекционного вируса при 4°С увеличился до 12 нед. Добавление MgS04 до концентрации 1 М облегчает очистку РСВ для физико-химических исследований, однако при выделении вируса и определении его инфекционной активности добавление MgS04 нежелательно, поскольку высокая ионная сила оказывает отрицательное воздействие на культуры клеток.
2.2 Методы определения инфекционности
Инфекционность препаратов РСВ можно определить методом бляшек. Метод, описываемый здесь для клеток BS-C-1, пригоден и для работы с большинством других клеточных культур.
1. Высевают по 106 клеток в чашки Петри диаметром 60 мм. Чашки инкубируют в течение ночи при 37 °С и используют клетки сразу же после образования плотного монослоя.
2. Культуральную среду удаляют и вносят в чашки по 0,2 мл последовательных 10-кратных разведений вирусного препарата. Для приготовления разведений можно использовать буферный раствор, например PBS, или, предпочтительно, среду MEM.

В обоих случаях используемый для разведения вируса раствор должен содержать 0,5% ЭТС и антибиотики.
3. Культуру инкубируют 1 ч при 31 °С для адсорбции вируса.
4. В каждую чашку вносят по 5 мл покрытия. Удаление инокулята необязательно. В качестве покрытия можно использовать среду MEM, содержащую 1-2% ЭТС, антибиотики и 0,9% агара. Эту среду готовят, смешивая равные объемы нагретой до 37 °С среды MEM в двойной концентрации и расплавленного 1,8% -ного агара Нобль. При температуре 43-46 °С агар не застывает, и покрытие можно наносить на слой клеток без их повреждения.
5. Клетки инкубируют 3-5 сут. при 31-39°С; время инкубации зависит от температуры: чем выше температура, тем короче время инкубации.
Для приготовления стабильных препаратов, в которых можно подсчитывать бляшки через длительное время после опыта, в каждую чашку добавляют 2 мл 1%-ного раствора глутарового альдегида в PBS и оставляют при комнатной температуре по крайней мере на 4 ч для фиксации. Фиксирующий раствор и агаровое покрытие удаляют и добавляют в чашки раствор подходящего красителя, окрашивают в течение 5 мин, затем тщательно промывают водой и сушат. Для максимально точного подсчета бляшек необходим бинокулярный микроскоп с небольшим увеличением. Для окрашивания можно использовать и 0,01%-ный раствор нейтрального красного в PBS или в среде, содержащей 0,9% агара.
Обычно для усиления контраста из состава агарового покрытия исключают феноловый красный, но это не имеет принципиального значения. Окрашивание раствором нейтрального красного проходит быстрее, однако такие препараты нестабильны и подвержены фотохимическому повреждению под действием света.
Окрашивание с помощью твердого нейтрального красного, входящего в состав агарового покрытия, происходит медленнее, однако в тех случаях, когда необходимо выделить инфекционный вирус из индивидуальных бляшек, данный метод явно предпочтительнее.
2.3 Гемагглютинация
В культуральной жидкости инфицированных РСВ клеток гемагглютинин не обнаруживается. Гемагглютинация с эритроцитами человека, африканской зеленой мартышки, макаки-резуса, морской свинки, крысы, хомячка, мыши, гуся, овцы, свиньи, курицы и цыпленка не отмечена.
С другой стороны, ВПМ агглютинирует эритроциты мыши и хомячка. Методические аспекты постановки реакции гемаглютинации описаны в работе Компанса и др.
2.4 Гемадсорбция
Адсорбция эритроцитов на клетках, зараженных РСВ, не описана. Негативные результаты получены с эритроцитами африканской зеленой мартышки, шимпанзе, обезьяны гелады, морской свинки, крысы, хомячка, мыши, утки, гуся, голубя, курицы и цыпленка. Однако клетки, зараженные ВПМ, связывают эритроциты мыши; технические детали описаны Ком-пансом и др. .
2.5 Иммунофлуоресцентные методы
Иммунофлуоресцентное окрашивание с помощью поликлональной антисыворотки представляет собой удобный и точный метод подсчета зараженных клеток и выявления места размножения вируса при анализе фиксированных срезов зараженных тканей. Используя моноклональные антитела против индивидуальных вирусных полипептидов, иммунофлуоресцентным методом можно изучать и внутриклеточную локализацию вирусных антигенов.
Иммунофлуоресцентное окрашивание применяется также для быстрой диагностики вирусных инфекций. Это направление недавно рассмотрено в исчерпывающих обзорах, а конкретные методы обнаружения РСВ в клиническом материале описаны в работе Гарднера и др. Иммунофлуоресценция - более точный метод выявления РСВ в организме больного, чем непосредственное выделение вируса, поскольку в период реконвалесценции после вызванного РСВ заболевания зараженные клетки бывают покрыты противовирусными антителами.
Непрямая иммунофлуоресценция более чувствительна, чем прямая, однако при ее использовании возникает проблема неспецифической флуоресценции. Ее можно устранить предварительным истощением антисыворотки и антииммуноглобулинового конъюгата обработанной ацетоном культурой клеток или гомогенатом печени животных. Обработка клеток ацетоном проводится следующим образом:
1. 10s-109 клеток снимают со стекла и получают суспензию.
2. Клетки концентрируют центрифугированием и ресуспендируют в 0,01 М PBS; эту процедуру повторяют трижды.
3. Осадок клеток ресуспендируют: 5 мин в 10-кратном объеме ацетона и вновь осаждают клетки.
4. Клетки ресуспендируют в 0,01 М PBS и проводят 6 циклов осаждения для полного удаления ацетона. После этого клетки можно хранить до употребления в 0,01 М PBS при - 20 °С.
Ниже приведена стандартная методика для выявления антигенов РСВ в зараженных клетках методом непрямой иммунофлуоресценции.
1. В пластиковые чашки диаметром 50 мм помещают тщательно вымытые и дегидратированные этанолом покровные стекла и высевают по ЫО6 клеток BS-C-1. Инкубируют клетки 24 ч, промывают и заражают РСВ с множественностью инфекции около 1 БОЕ/кл.
2. После адсорбции вируса в течение 2 ч покровные стекла с зараженными клетками тщательно промывают средой MEM, содержащей 2,5% ЭТС, и инкубируют при 31 или 39 °С.
3. Через 40-48 ч после заражения клетки фиксируют на 5 мин в охлажденном до 4°С ацетоне, высушивают на воздухе 30 мин и хранят при - 20 °С.
4. Антивирусную сыворотку истощают обработанными ацетоном клетками BS-C-1 при 4°С и используют в подобранном оптимальном разведении. Кроличья сыворотка против глобулинов нескольких видов животных, конъюгированная с изотиоцианатом флуоресцеина, имеется в продаже. Конъюгат истощают обработанным ацетоном гомогенатом печени мыши или клетками BS-C-1 при 4°С.
5. Покровные стекла инкубируют с антивирусной сывороткой 1 ч при 37 °С, промывают и инкубируют еще 45 мин при 37 °С с соответствующим конъюгатом.
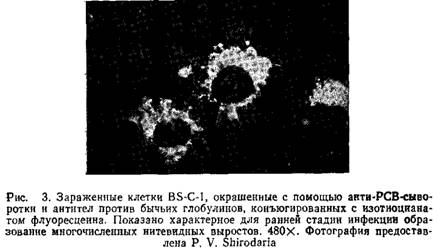
6. После промывки препараты заключают в забуференный глицерин и исследуют под флуоресцентным микроскопом. Рис.3 иллюстрирует разрешение, которое может быть получено при использовании данного метода.
Доказательствами специфичности иммунофлуоресценции могут служить:
1) отсутствие флуоресценции незараженных клеток BS-C-1;
2) отсутствие флуоресценции при использовании ЭТС вместо специфической с зараженными РСВ клетками;
3) отсутствие флуоресценции после истощения противовирусной сыворотки зараженными клетками.
Высокой чувствительностью обладает и иммунопероксидазный метод; его преимущество состоит в том, что он не требует использования микроскопа с ультрафиолетовой оптикой.
2.6 Твердофазный иммуноферментный анализ
ELISA - еще один иммунологический метод быстрого обнаружения и количественного определения РСВ. Ниже приведена типичная методика.
1. Лунки планшета для иммунологических реакций покрывают зараженными или незараженными клетками, или РСВ, очищенным центрифугированием в градиенте. Это делается одним из следующих методов:
а) клетки выращивают в лунках планшета и через ряд заражают РСВ. Когда в лунках с зараженными клетками проявляется ЦПД, клетки промывают, фиксируют раствором, содержащим этанол и уксусную кислоту, и хранят при - 20 °С;
б) в лунки планшета вносят РСВ, очищенный центрифугированием в градиенте, и высушивают в течение ночи при 37°С.
Если имеются клетки, персистентно инфицированные РСВ, их можно использовать в качестве источника антигена вместо литически инфицированных клеток.
2. Перед началом анализа все лунки планшета покрывают 5% -ным раствором БСА в PBS. В каждый опыт следует включать негативный контроль и позитивный контроль. В лунки планшета вносят образцы вируса и инкубируют в течение ночи при 4°С. Планшет промывают и добавляют овечьи антитела против иммуноглобулинов мыши, конъюгированные с пероксидазой хрена в рекомендуемом разведении. Планшет инкубируют 1 ч при 37 °С, промывают, добавляют субстрат, о-фенилендиамин, и инкубируют при 37 °С еще 30 мин. Реакцию останавливают добавлением 4,5 М H2SO4 и учитывают результаты с помощью ридера "Мультискан".
Хиерхольцер и др. описали методику, позволяющую с помощью ELISA количественно выявлять белковые полосы после электрофореза и переноса белков на нитроцеллюлозные мембраны, так называемый метод "вестерн-блотинга". Преимущество этого метода состоит в том, что он не требует использования радиоактивных изотопов.
3. Аналитические методы
3.1 Радиоактивное лечение, радиоиммуноанализ и радиоиммунопреципитация
3.1.1 Введение радиоактивной метки в вирусные белки в зараженных РСВ клетках
1. Оптимальные условия для мечения вирусных белков достигаются при добавлении к зараженным клеткам через 18 ч после заражения актиномицина D в концентрации 2,5 мкг/мл.
Белки РСВ удается эффективно пометить смесью - Meтионина и - цистеина по следующей методике.
2. Через 2 ч после внесения актиномицина D культур ал ьную среду заменяют на среду, содержащую 50 мкКи/мл L--Meтионина и 25 мкКи/мл Ь--цистеина. у
3. Клетки инкубируют 16 ч при 37 °С или до проявления выраженного ЦПД.
4. Монослой промывают, солюбилизируют клетки и экстрагируют белки лизирующим буфером.
5. После инкубации в течение 2 ч при комнатной температуре лизат используют для электрофореза или хранят при - 20 °С.
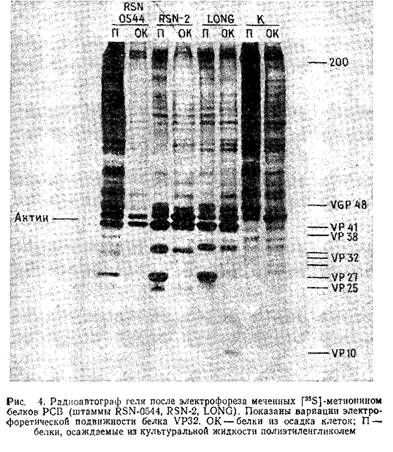
Рис.4 иллюстрирует результаты разделения меченных - метионином белков РСВ в пластине 10% -ного полиакриламидного геля. Вирусные гликопротеины можно пометить аналогичным методом, используя в качестве метки 50 мкКи/мл - глюкозамина или - маннозы. Альтернативный метод введения метки в гликопротеины оболочки РСВ заключается в йодировании поверхности зараженных вирусом клеток лактопероксидазным методом. Белки очищенного нуклеокапсида РСВ можно йодировать с помощью хлорамина Т.
3.1.2 Радиоиммуноанализ
Метод прямого RIA с использованием в качестве вторых антител меченных 1251 гамма-глобулинов сыворотки козы против глобулинов мыши, а в качестве субстрата - фиксированных метанолом персистентно инфицированных РСВ клеток описан Коутом и др. .
3.1.3 Радиоиммунопреципитация
Радиоиммунопреципитация проводится стандартным методом, описанным, например, Ферни и Герином и Уордом и др. Для РСВ применяют следующую методику.
1. "Монослойные культуры клеток в чашках Петри диаметром 50 мм заражают РСВ с множественностью инфекции более 1 БОЕ/кл. Через определенные промежутки времени после заражения проводят импульсное мечение. Для этого культуральную среду в чашках заменяют на 1 мл PBS, содержащего 30 мкКи - метионина и 25 мкКи - цистеина, и инкубируют чашки при 31 °С 1 ч.
2. Радиоактивный раствор сливают и монослой промывают охлажденным PBS.
3. Клетки снимают со стекла с помощью стерильной резиновой палочки и осаждают центрифугированием.
4. Клетки ресуспендируют в 200 мкл лизирующего буфера, инкубируют суспензию 30 мин в ледяной бане, затем интенсивно перемешивают.
5. Ядра и клеточный дебрис удаляют центрифугированием 5 мин при 10 000 g.
6. Лизат осветляют инкубацией с 50 мкл 10% -ной суспензии фиксированных формалином стафилококков или с 50 мкл суспензии покрытых белком-А гранул 30 мин при 0°С.
7. Стафилококки удаляют центрифугированием. К 50 мкл лизата добавляют 10 мкл гипериммунной сыворотки, перемешивают и инкубируют 3 ч во льду.
8. Добавляют 100 мкл суспензии стафилококков, перемешивают и инкубируют смесь 30 мин при комнатной температуре.
9. Иммунные комплексы осаждают центрифугированием 20 с при 10 000 g.
10. Осадок 3 раза промывают раствором, содержащим 500 мМ LiCl, 100 мМ трис-HCl, рН 8,5.
11. Осадок после промывки ресуспендируют в 50 мкл смеыг для диссоциации, кипятят 2 мин и либо немедленно наносят на гель для электрофореза, либо хранят при - 20 °С.
3.2 Метод выявления дефектных интерферирующих частиц
Дефектные интерферирующие частицы и стандартные вирионы РСВ не удается разделить физическими методами, на проявление устойчивой к ультрафиолетовому облучению и чувствительной к нейтрализующим антителам интерферирующей активности при пассировании вируса с высокой множественностью инфекции свидетельствует о накоплении ДИЧ. Разра-Зотзн колориметрический метод количественного определения ДИЧ в препаратах РСВ. Он основан на измерении интенсивности окрашивания, обусловленного поглощением нейтрального красного клетками, которые выживают при заражении стандартным вирусом в результате защиты ДИЧ. Приводятся данные о том, что колориметрический метод чувствительнее, чем метод, основанный на определении снижения выхода вируса. Колориметрический анализ проводят следующим образом.
1. Монослойные культуры клеток Нер-2, выращенные в лунках планшета диаметром 16 мм, инкубируют с содержащим ДИЧ материалом 2 ч при 37 °С.
2. Содержащий ДИЧ иннокулят удаляют и заражают клетки стандартным вирусом, полученным в результате пассирования с низкой множественностью, и продолжают адсорбцию 2 ч при 37 °С.
3. В каждую лунку вносят по 1 мл культуральной среды и инкубируют клетки 72 ч при 37 °С.
4. Среду удаляют и вносят в каждую лунку по 0,5 мл нейтрального красного в растворе Эрла; инкубацию клеток продолжают еще 2 ч при 37 °С.
5. Краситель удаляют и клетки дважды промывают PBS.
6. Краситель экстрагируют из клеток 1 мл 50% -ного этанола, содержащим 0,1 М NaH2P04, и определяют его количество спектрофотометрически при 540 нм. Полученные данные сравнивают с количеством красителя, экстрагированного из незараженных клеток и из клеток, зараженных стандартным вирусом.
Концентрацию ДИЧ можно рассчитать следующим образом, предполагая, что их распределение по клеткам подчиняется закону Пуассона:
число ДИЧ/мл= 1/разведение, обеспечивающее выживание 63% клеток Х общее число клеток X 1/объем иннокулята.
3.3 Электронная микроскопия
Пневмовирусы исключительно многообразны по форме; кроме того, их вирионы и нуклеокапсиды нестабильны и легко повреждаются при обычных процедурах концентрирования вирусов. Поэтому точный подсчет вирусных частиц практически невозможен.
Трансмиссионная электронная микроскопия удобна для идентификации вирусов и изучения морфологии и морфогенеза ви-рионов. Сканирующая электронная микроскопия используется для изучения морфологии поверхности зараженных клеток, а также для количественного определения поверхностных антигенов на основе специфического связывания бактериальных клеток.
3.3.1 Морфология вирионов
Для негативного контрастирования успешно применяют следующие вещества: фосфорно-вольфрамовая кислота, фосфовольфрамат натрия, фосфовольфрамат калия, кремневольфрамат натрия и уранилацетат. Концентрирование вируса необходимо проводить самым мягким методом, например осаждением полиэтиленгликолем или ультрафильтрацией через фильтры "Амикон", избегая высокоскоростного центрифугирования. Каплю концентрированного раствора вируса наносят на покрытую графитом формваровую сеточку. Для фиксации добавляют равный объем 3% -ного глутарового альдегида в PBS, перемешивают и инкубируют 5 мин при комнатной температуре. Сеточки промывают PBS и проводят негативное контрастирование. Для визуализации нуклеокапсидов зараженные клетки лизируют дистиллированной водой или гипотоническим буферным раствором. Клеточный дебрис можно осадить непосредственно на сеточке перед негативным контрастированием.
3.3.2 Морфогенез вирионов
1. Зараженные клетки фиксируют, добавляя к монослою, выращенному в чашке Петри диаметром 50 мм, глутаровый. альдегид до концентрации 2,5%.
2. После фиксации в течение 30 мин монослой дважды промывают PBS и добавляют 10 капель 1% -ного раствора четы-рехокиси осмия.
3. Через 15 мин монослой еще 2 раза промывают PBS, снимают клетки со стекла и суспендируют их в 2 мл PBS.
4. Клетки осаждают центрифугированием и дегидратируют, последовательно суспендируя в растворах с возрастающей концентрацией этанола; в каждом растворе клетки инкубируют 5 мин; при необходимости их можно выдержать в течение ночи в 70% -ном этаноле. После инкубации в 90% -ном этаноле клетки дважды по 15 мин обрабатывают 100% -ным этанолом.
5. Этанол заменяют на смесь этанола и окиси пропилена.
6. Через 15 мин смесь этанол-окись пропилена заменяют на свежую, инкубируют суспензию еще 30 мин, после этого ресуспендируют клетки в смеси этанол-окись пропилена-среда EPON, а затем в среде EPON 1 ч.
7. Компактный осадок клеток наносят на небольшой объем среды EPON в капсуле ВЕЕМ и, заполнив капсулу средой EPON, инкубируют их 48 ч при 60 °С для полимеризации. После этого заключенные в капсулу клетки готовы для приготовления срезов стандартным способом на ультрамикротоме.
8. Тонкие срезы располагают на необработанных сеточках для электронной микроскопии и через 24 ч окрашивают 5 мин,, помещая сеточки препаратами вниз в каплю насыщенного раствора уранилацетата в метаноле.
9. Сеточки промывают дистиллированной водой и сушат на листе фильтровальной бумаги.
10. На срез наносят каплю 0,1 М раствора NaOH и помещают сеточку срезом вниз на 5 мин в каплю раствора ацетата свинца. Сеточки промывают и сушат на листе фильтровальной бумаги, после этого они готовы к электронно-микроскопическому исследованию.
3.3.3 Сканирующая электронная микроскопия
Для подготовки клеток к сканирующей электронной микроскопии применяют следующий метод.
1. Клетки, образующие неплотный монослой на покровных стеклах, фиксируют 1-2 ч 2,5% -ным раствором глутарового альдегида в фосфатном буфере.
2. Стекла помещают на 1 ч в 1% -ный раствор четырехокиси осмия в фосфатном буфере.
3. Клетки дегидратируют, последовательно помещая стекла в 30, 50, 70, 90 и 100% -ный этанол, а затем на 15 мин в смесь этанола с ацетоном.
4. Стекла дважды обрабатывают 100% -ным ацетоном и сушат С02 в критической точке, используя аппарат для сушки фирмы Polaran Equipment Ltd., Watford, Herts или другой аналогичный прибор.
5. Покровные стекла фиксируют на алюминиевых подложках с помощью клея "Electrocday 915" и покрывают золотом с помощью диодного напылителя Polaron Е5000 или другой аналогичной системы. Подготовленные таким образом препараты можно исследовать методом сканирующей электронной микроскопии.
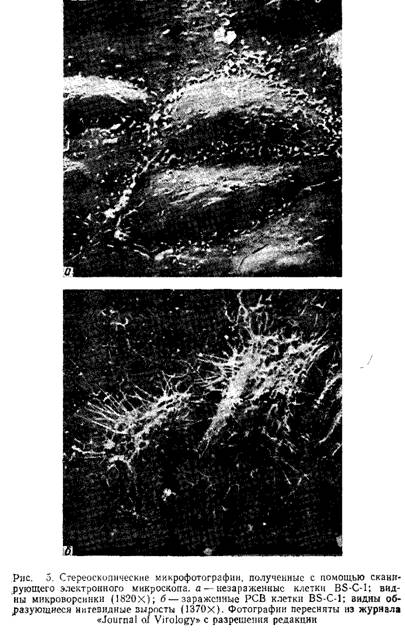
На рис.5 представлена фотография зараженных пневмо-вирусом клеток, выполненная с помощью сканирующей электронной микроскопии. Обратите внимание на длинные выросты инфицированных клеток.
3.3.4 Локализация и количественное определение вирусных антигенов по связыванию стафилококков
Клетки Staphylococcus aureus с помощью поверхностного А-белка связываются с Fc-фрагментами иммуноглобулинов. Этот эффект можно использовать для исследования пространственной локализации вирусного антигена на плазматической мембране зараженных клеток после их обработки противовирусной сывороткой. Стафилококки более удобны, чем покрытые А-белком шарики латекса, полиметил-метакрилата или полистирола, в связи с легкостью получения бактерий и одинаковым диаметром всех бактериальных клеток (около 1 мкм). На поверхности клеток, зараженных многими вирусами, стафилококки распределяются равномерно, но в случае пневмовирусов они специфически связываются с нитевидными выростами, образование которых является уникальной особенностью зараженных этими вирусами клеток. Связывание стафилококков высокоспецифично и подтверждает результаты иммунофлуоресцентного окрашивания, которые свидетельствуют о том, что вирусные антигены на поверхности зараженных клеток локализованы преимущественно в составе нитевидных структур.
Связывание стафилококков с поверхностью зараженных клеток проводят по следующей методике.
1. Клетки выращивают на покровных стеклах до образования монослоя и заражают вирусом. Лучше всего использовать клетки почки кенгуровой крысы, поскольку они высокочувствительны к пневмовирусам и не имеют поверхностных выростов.
2. Зараженные клетки инкубируют в поддерживающей среде, промывают PBS для полного удаления входящей в состав среды сыворотки, затем фиксируют 10-15 мин 2,5% -ным раствором глутарового альдегида в PBS при комнатной температуре.
3. Клетки обрабатывают противовирусной сывороткой 1 ч при 37 °С.
4. Антисыворотку удаляют, тщательно промывая клетки PBS. К клеткам добавляют суспензию aureus, штамм Cowan, приготовленную по методу Прингла и Парри, или коммерческий препарат стафилококков и инкубируют 5 мин при 37 °С.
5. Несвязавшиеся стафилококки удаляют, быстро промывая стекла PBS, и клетки фиксируют 60 мин при комнатной температуре 2% -ным раствором четырехокиси осмия в PBS. После этого стекла можно подготовить к сканирующей электронной микроскопии и исследовать.
Подсчет числа связавшихся с клетками стафилококков дает возможность количественно исследовать экспрессию вирусных антигенов на клеточной мембране при условии постановки соответствующих контролей.
3.4 Концентрирование вируса
Предложен простой метод концентрирования РСВ преципитацией полиэтиленгликолем.
1. Готовят 36% -ный раствор ПЭГ 6000 в дистиллированной воде.
2. Раствор охлаждают и добавляют к содержащей вирус жидкости до конечной концентрации ПЭГ 6%.
3. Суспензию инкубируют 2 ч при 4°С.
4. Преципитат собирают центрифугированием при 4°С.
5. Супернатант сливают, осадок дважды промывают буферным раствором и ресуспендируют в небольшом объеме.
3.5 Очистка и радиоактивное мечение РСВ
Клинические изоляты РСВ в основном ассоциированы с клетками, однако при культивировании некоторых лабораторных штаммов в среду выделяется достаточное количество вируса для успешной очистки. Наиболее удобно использовать для этого штаммы Лонг и А2.
3.5.1 Градиентное центрифугирование
В литературе можно встретить не одну методику очистки РСВ градиентным центрифугированием. Представленная ниже была успешно использована для очистки штамма А2.
1. Монослой клеток Нер-2 заражают РСВ. Адсорбцию проводят 2 ч при 37°С, затем добавляют культуральную среду.
2. Через 10 ч после заражения добавляют актиномицин D до концентрации 0,3 мкг/мл. Для радиоактивного мечения РНК через 16-20 ч после заражения заменяют культуральную среду на свежую, содержащую 20 мкКи/мл - уридина. Для введения метки в белки в среду добавляют 5 мкКи/мл - метионина.
3. При проявлении ЦПД и образовании синцитиев собирают культуральную жидкость. Клеточный дебрис удаляют центрифугированием 20 мин при 11000 g и концентрируют вирус центрифугированием 90 мин при 65000 g на холоду.
4. Осадок ресуспендируют в буфере NTE при ультразвуковой обработке.
5. Вирус центрифугируют 1 ч при 150 000 g в 10-50% -ном градиенте концентрации сахарозы и собирают фракции, содержащие видимую зону вируса.
6. Содержащие вирус фракции объединяют, разводят NTE и обрабатывают ультразвуком. Вирус вновь центрифугируют 2 ч при 150 000 g в 20-60% -ном градиенте концентрации сахарозы.
7. Вирус концентрируют повторным центрифугированием 90 мин при 65000 g.
Данная методика позволяет получить частично очищенные препараты радиоактивно меченного РСВ. Для последующей очистки предложены более сложные методы; инфекционность, которая в обычных условиях очень чувствительна к центрифугированию, можно стабилизировать добавлением MgS04 до концентрации 1 М.
3.5.2 Очистка РСВ методом изопикнического центрифугирования
Данный метод требует более длительного центрифугирования в градиенте концентрации сахарозы или метризамида. Растворы метризамида обладают более низкой вязкостью и осмотическим давлением, чем растворы сахарозы, и поэтому их использование для изопикнического центрифугирования предпочтительно. Растворы метризамида всегда следует готовить непосредственно перед использованием на 10 мМ HEPES, рН 7,8.
3.6 Радиоактивное мечение вирионной РНК
Интактную вирионную РНК РСВ получить трудно; рекомендуется следующая методика.
Очищенный радиоактивно меченный вирус получают, РНК экстрагируют фенолом и далее из водной фазы осаждают этанолом.
Выделенную РНК очищают центрифугированием в 15 - 30% -ном градиенте концентрации сахарозы в присутствии ДДС-Na в течение 15 ч при 19 000 об/мин.
Альтернативный подход состоит в том, что РНК, экстрагированную из немеченых вирионов, метят по З'-концу с помощью РНК-лигазы и - цитидин-3/,5'-бисфосфата, как описано Вертц и Дэвисом. РНК можно пометить и по 5'-концу после удаления 5'-концевого фосфата щелочной фосфатазой из кишечника крупного рогатого скота, которую затем инактивируют протеиназой К. После этого РНК экстрагируют фенолом, осаждают этанолом и метят в присутствии - ATP и полинуклеотидкиназы из Е. coli, зараженной бактериофагом Т4. Меченую РНК затем отделяют от непрореагировавшего АТР хроматографией на колонке с сефадексом G50. Методика, удобная для мечения РНК РСВ, описана Шубертом и др. .
3.7 Анализ вирусных РНК и белков методом электрофореза в полиакриламидном геле
3.7.1 Анализ белков
Синтезированные in vivo или in vitro меченые белки можно анализировать с помощью электрофореза в 10% -ном или б - 15% -ном градиентном полиакриламидном геле, как описано Марсденом и др. .
После электрофоретического разделения в полиакриламидном геле белки можно перенести на нитроцеллюлозный фильтр для биохимического или иммунологического анализа. Применение этого метода к белкам РСВ описано Хиерхольцером и др. Перенос белков из. полиакриламидного геля на нитроцеллюлозу осуществляется электрофоретически по нижеследующей методике.
1. После электрофоретического разделения белков полиак-риламидный гель помещают в камеру прибора для электрофоретического переноса вместе с листом нитроцеллюлозы. В камеру заливают буферный раствор, содержащий 0,025 М трис, 0, 193 М глицин и 20% метанола, рН 8,35.
2. Для переноса проводят электрофорез при 60 В по крайней мере 4 ч при 4°С.
3. Лист нитроцеллюлозы разрезают на полоски и помещают каждую в закрытую пробирку.
4. Полоски инкубируют с анти-РСВ сывороткой 2 ч при комнатной температуре.
5. Полоски 3 раза промывают PBS, содержащим твин 80.
6. Полоски инкубируют с конъюгатом, разведенным в 1000 раз, 3 ч при комнатной температуре.
7. Полоски 3 раза промывают тем же раствором.
8. Нитроцеллюлозу проявляют раствором, содержащим 50 мг 3,3'-диаминобензидина и 100 мкл 3% -ной перекиси водорода в 100 мл PBS, рН 7,2.
9. Нитроцеллюлозу промывают водой и сушат на листе фильтровальной бумаги.
10. Количественный анализ можно провести спектрофотометрически после обработки нитроцеллюлозы веществами, растворяющими пластмассы. В результате подобной обработки нитроцеллюлоза становится прозрачной.
3.7.2 Анализ РНК
Радиоактивно меченную РНК из очищенных вирионов или из цитоплазмы зараженных РСВ клеток можно проанализировать электрофорезом в 1,5% -ном геле агарозы, содержащем 6 М мочевину, по следующей методике.
1. Для приготовления геля размером 50X17X0,3 см готовят отдельно следующие растворы: а) 120 мл 4,5% -ной агарозы; для растворения агарозы суспензию обрабатывают в микроволновой печи или автоклавируют ее; б) 1,5-кратный концентрат раствора мочевины в цитратном буфере; для этого к 129,6 г мочевины добавляют 9 мл концентрированного 1 М цитратного буфера и растворяют, доводя объем водой до 240 мл.
2.1,5-кратный буфер нагревают и смешивают с 3-кратным концентратом агарозы.
3. Гель заливают на холоду в горизонтальный аппарат для электрофореза в 3 стадии, перерывы между стадиями составляют 45 мин.
4. В гель немедленно вводят гребенку.
5. Гель используют для электрофореза через 2-3 ч после заливки. Гребенку вынимают, промывают ячейки буфером с мочевиной и заполняют их тем же буфером. В ячейки вносят образцы, смешанные с буфером для нанесения.
6. Электрофорез проводят на холоду при градиенте напряжения 5 В/см в течение 18 ч. Основные растворы готовят следующим образом.
1 М цитратный буфер, рН 3,0: смешивают 1 М растворы лимонной кислоты и цитрата натрия до достижения рН 3,0. Буфер для электрофореза: 1 М. цитратный буфер разводят в 40 раз, получая 0,025 М цитратный буфер, рН 3,0 Буфер для нанесения образцов: 0,025 мл 1 М цитратного буфера, 3,6 г 6 М мочевины, 2 г 20% -ной сахарозы, 0,1 мл 0,005% -ного бромфенолового синего, объем доводят водой до 10 мл. Буфер с 6 М мочевиной: 3,6 г мочевины, 0,25 мл 1 М цитрата, рН 3,0; объем доводят до 10 мл дистиллированной водой.
3.8 Очистка матричной РНК для трансляции
Матричную РНК для трансляции in vitro получают следующим образом.
1. 50-Ю6 клеток заражают РСВ. Клетки инкубируют 10-12 ч при 37 °С.
2. Монослой промывают, снимают клетки со стекла и суспендируют в буферном растворе. Суспензию оставляют на 10 мин при 4°С для набухания клеток.
3. Клетки разрушают в гомогенизаторе Даунса, затем осаждают клеточный и дебрис центрифугированием при 4°С.;
4. Осадок ресуспендируют в 2 мл того же буфера, вновь гомогенизируют и центрифугируют. Супернатанты, полученные после первого и второго центрифугирований, объединяют.
5. К супернатанту добавляют CsCl и N-лаурилсаркозин. Раствор тщательно перемешивают и прогревают 1-2 мин при 51 °С.
6. 7 мл полученного раствора наслаивают на 2 мл раствора в пробирке для ротора SW40 и добавляют сверху буфер, чтобы заполнить пробирку. Пробирки центрифугируют в роторе SW40 16 ч при 25000 об/мин и 25 °С.
7. Осадок ресуспендируют в 0,5 мл стерильной дистиллированной воды. РНК дважды переосаждают этанолом, добавляя 1,2 мл этанола, 25 мкл 4 М раствора NaCl и 10 мкл 10% -ного ДДС-Na.
8. После второго осаждения осадок ресуспендируют в буфере, добавляют ДДС-Na до концентрации 0,2% и проводят хроматографию на колонке с олиго - целлюлозой. Связавшийся материал элюируют и осаждают РНК этанолом, добавляя в ка* честве носителя тРНК из печени кролика. Осадок ресуспендируют в том же буфере без ДДС-Na и переосаждают РНК двумя объемами этанола в присутствии 0,2 М ацетата калия. Конечный осадок ресуспендируют в 250-500 мкл 0,01 М HEPES, рН 7,6. В качестве бесклеточной системы для синтеза белка можно использовать зависимый от мРНК лизат ретикулоцитов кролика, приготовленный, как описано Престоном, или коммерческий препарат лизата. Для контроля следует параллельно получить мРНК из незараженных клеток. Индивидуальные вирусные мРНК для трансляции в бесклеточной системе можно выделить методом гибридизационной селекции с использованием клонов кДНК с известной структурой, как описано Коллинзом и Вертц.
3.9 Молекулярное клонирование и олигонуклеотидное секвенирование
10 генов РСВ были идентифицированы в результате клонирования кДНК с последующим картированием и трансляцией вирусных мРНК - кДНК получали обратной транскрипцией мРНК из зараженных РСВ клеток Нер-2 и клонировали их в Е. coli с использованием плазмиды pBR322.
Нуклеотидная последовательность гена белка нуклеопротеи-на РСВ и кодируемая ею аминокислотная последовательность - опубликованы Венкатесаном и Эланго, а последовательности гена и белка М. - Сатаке и Венкатесаном. В этих работах подробно описаны методы олигонуклеотидного секвенирования в применении к РСВ.
3.10 Очистка вирусных белков
3.10.1 Вирусные полипептиды
В настоящее время очищенные вирусные белки можно получать методом аффинной хроматографии с использованием специфических моноклональных антител. Другой подход состоит в экспрессии клонированных генов в прокариотических или в эукариотических клетках. Однако непосредственно для РСВ такие методики пока не описаны.
3.10.2 Очистка капсидного белка
Капсидный белок выделяют из очищенных нуклеокапсидов, которые в свою очередь можно получить либо из вирионов, либо из экстракта зараженных клеток. Ниже приведена методика выделения из клеточных экстрактов, дающая более высокий выход.
50-Ю6 клеток заражают РСВ. Через 36-48 ч после заражения клетки снимают с поверхности флакона раствором, содержащим 10 мМ трис-НО, рН 7,4, 0,18 М NaCl, 0,25 мМ ЭДТА и 0,1 мМ PMSF. Клетки осаждают центрифугированием и ресуспендируют в 1мл лизирующего буфера 20 мин при 4°С. Клетки разрушают в гомогенизаторе Даунса. К гомогенату добавляют NaCl до концентрации 0,1 М и удаляют неразрушенные клетки, ядра и дебрис центрифугированием 2 мин при 600 g. Нуклеокапсиды осаждают центрифугированием 30 мин при 100000 g. Нуклеокапсиды промывают лизирующим буфером, а затем буфером, содержащим 10 мМ трис-HCl, рН 7,4, и 0,1 мМ ЭДТА. Осадок нуклеокапсидов ресуспендируют и получают очищенные нуклеокапсиды изопикническим центрифугированием в 15-55% -ном градиенте концентрации тартрата калия в буфере ТЕ.
3.10.3 Очистка вирионных гликопротеинов
Предложен метод очистки гликопротеинов РСВ, основанный на солюбилизации вирионов неионным детергентом в буферном растворе с низкой ионной силой.
1. Очищенные методом градиентного центрифугирования вирионы осаждают центрифугированием через слой 30% -ного раствора сахарозы в буфере NTE при 50 000 g 60 мин. Осадок ресуспендируют в 0,01 М фосфатном буфере, содержащем 10% тритона Х-100.
2. Суспензию перемешивают 30 мин при комнатной температуре и центрифугируют 60 мин при 200 000 g.
3. Супернатант экстрагируют п-бутанолом.
4. Экстрагированные белки суспендируют в 0,01 М фосфатном буфере с 1% тритона Х-100 и фракционируют в 5-25% -ном линейном градиенте концентрации сахарозы, содержащем 1% тритона Х-100, 18 ч при 100 000 g. Собирают фракции из верхней части градиента и диализуют их против 0,01 М фосфатнога буфера.
5. Тритон Х-100 удаляют экстракцией п-бутанолом.
6. Фракции, содержащие вирусные гликопротеины, идентифицируют с помощью электрофореза в полиакриламидном геле.
4. Специальные биологические методы
4.1 Получение моноклональных антител
Моноклональные антитела к нескольким белкам РСВ были получены стандартными методами. Ниже приведен типичный протокол.
1. Мышам линии Balb/c внутрибрюшинно вводят зараженные РСВ клетки. Через 10 сут. инокуляцию повторяют.
2. Селезенки иммунизированных мышей суспендируют в среде RPM1 1640, забуференной бикарбонатом натрия и содержащей пенициллин, гентамицин, пируват натрия, L-глютамин и ЭТС.
3. Клетки селезенки иммунизированных мышей сливают с клетками мышиной миеломы, например Balb/c NS1/1, выращиваемой в той же среде. При этом смешивают 3-107 клеток селезенки с таким же количеством клеток миеломы. Клетки центрифугируют 10 мин при 200 g и 1 мин выдерживают при 37 °С. Для слияния клеток по каплям добавляют 1 мл 50% -ного ПЭГ 4000 с 5% ДМСО.
4. Суспензию инкубируют 90 с при 37 °С и останавливают процесс слияния, добавляя 20 мл среды.
5. Клетки осаждают центрифугированием и промывают средой. Осадок ресуспендируют в среде ГАТ и распределяют по плоскодонным лункам планшета для иммунологических реакций.
6. Клетки инкубируют на питательном слое из макрофагов, подготовленном за сутки до слияния; каждая лунка должна содержать 10" перитонеальных макрофагов здоровой мыши.
7. Через 6 сут после начала инкубации в каждую лунку вносят по 50 мкл среды ГАТ.
8. Через 9-12 сут после слияния проверяют активность и специфичность продуцируемых антител методом ELISA с антигенами РСВ.
9. Гибридомы, продуцирующие специфические антитела, клонируют методом предельного разведения в планшете для иммунологических реакций, используя макрофаги или клетки селезенки в качестве фидера. Клонирование проводят дважды. Гибридомы считают стабильными, если 95% клонов продуцируют специфические антитела.
4.2 Выделение температурно-чувствительных мутантов
Для анализа функций генов и биохимических исследований полезно иметь температурно-чувствительные мутанты вируса. Их выделяют следующим образом.
1.106 клеток BS-C-1, выращенных в чашке Петри диаметром 50 мм или во флаконе с завинчивающейся крышкой емкостью 30 мл, заражают РСВ дикого типа с множественностью инфекции 0,01 БОЕ/кл. Адсорбцию проводят 1 ч при 31 °С.
2. Вирус отмывают двумя сменами среды и добавляют 3 мл среды Игла, содержащей 2,5% ЭТС и мутаген.
3. Культуру инкубируют при 31 °С до проявления ЦПД в контрольной зараженной культуре, к которой мутаген не добавляли.
4. Разведения культуральной жидкости из обработанных мутагеном культур титруют методом бляшек после осветления, но без замораживания и оттаивания. Для этого на клетки наносят необходимые разведения и инкубируют клетки, под 0,9% -ным агаровым покрытием 5-7 сут при 31 °С.
5. Вирус из полученных индивидуальных бляшек наращивают, перенося бляшки пастеровской пипеткой на свежие монослойные культуры.
6. Проводят скрининг полученных изолятов вируса на способность давать бляшки при 31 и 39 °С. Для этого зараженные чашки инкубируют параллельно при каждой температуре. Альтернативный метод состоит в прямом скрининге бляшек, образованных вирусом, обработанным мутагеном. Для этого 2 чашки с культурой, покрытой слоем 0,9% -ного агара толщиной 2 мм, делят на секторы и в каждый сектор помещают 1 бляшку., После адсорбции в течение 30 мин при 31 °С заливают второй слой агарового покрытия. После этого одну чашку инкубируют при 31 °С, а другую при 39 °С. Из секторов, где бляшки образуются при 31 °С, но не при 39 °С, их извлекают для дополнительной проверки.
Температурно-чувствительные мутанты можно подразделить на функциональные группы с помощью теста комплементации, факторы, влияющие на комплементацию, детально изучены.
Как и для остальных вирусов с негативной полярностью РНК и несегментированным геномом, рекомбинация между температурно-чувствительными мутантами РСВ, принадлежащими к одной или к разным группам комплементации, не описана.
4.3 Получение персистентно инфицированных клеточных культур
Персистентной инфекции культур клеток при полном отсутствии ЦПД или минимальном его проявлении можно добиться несколькими способами. Одним из них является пассирование вируса с высокой множественностью инфекции и пересев выживающих клеток. Другие методы включают заражение частично устойчивых клеток в присутствии противовирусных антител или интерферона. Для получения персистентно инфицированных культур можно инкубировать клетки, зараженные температурно-чувствительными мутантами РСВ при частично ограничивающей размножение температуре для подавления ЦПД. Такие культуры после установления персистенции поддерживают при той же температуре. Исход опыта зависит от конкретных свойств используемого мутанта. Температурно-чувствительные мутанты комплементационной группы В, продуцирующие при неразрешающей температуре лишь небольшое количество антигена, не подходят для данной цели, поскольку дают либо абортивную, либо литическую инфекцию. С другой стороны, мутанты группы D, синтезирующие при неразрешающей температуре значительное количество антигена, воспроизводимо дают персистентно инфицированные культуры, которые можно пассировать неограниченное число раз. Персистентно инфицированные культуры служат хорошим источником вирусного антигена для проведения анализа по методу ELISA.