
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Контрольная работа: Методы контроля и анализа веществ (химические методы)
Контрольная работа: Методы контроля и анализа веществ (химические методы)
Контрольная работа № 1
МЕТОДЫ КОНТРОЛЯ И АНАЛИЗА ВЕЩЕСТВ (ХИМИЧЕСКИЕ МЕТОДЫ)
1. (1,10) Качественная реакция на кобальт (Со)
Решение:
Кобальт можно идентифицировать, переведя его из пробы в раствор в виде ионов Co2+ по реакции
3Co + 8HNO3 (разбавленная, горячая) = 3Co(NO3)2 + 2NO↑ + 4H2O
После этого можно проводить качественные реакции на ионы Co2+.
1) Действие реагента Ильинского (1-нитрозо-2-нафтола):
В реакции Co2+ с реагентом Ильинского при участии растворимого кислорода получается нерастворимое темно-красное хелатное соединение кобальта (III):
Co2+ + 12C10H6(NO)OH + O2 = 4[C10H6(NO)O]3Co↓ + 2H2O + 8H+
Строение комплексного соединения такое:
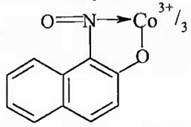
Осадок [C10H6(NO)O]3Co не растворяется в кислотах и щелочах. Идентификацию Co2+ проводят в кислой среде. Мешают ионы Cu2+, Fe3+, Fe2+.
2) Качественная реакция на ионы Co2+ с образованием кобальтонитрита (III) калия:
Нитрит калия образует с ионами Co2+ в уксуснокислом растворе при pH 4 – 5 желтый кристаллический осадок состава K3[Co(NO2)6]. Образованию осадка предшествует окисление Co2+ нитритом:
Co2+ + NO2- + 2H+ = Co3+ + NO↑ + H2O
Суммарный процесс можно показать уравнением:
Co2+ + 7NO2- + 3K+ + 2H+ = K3[Co(NO2)6]↓ + NO↑ + H2O
В разбавленных растворах осадок образуется при нагревании и отстаивании. Реакцию часто используют для отделения кобальта от мешающих ионов. Граница определения кобальта – 0,4 мкг.
2. (2,10-11) Определить нормальность раствора Ca(OH)2, титр которого составляет 0,00568 г/мл
Решение:
![]()
где ![]() – титр раствора,
– титр раствора,
![]() – нормальность раствора,
– нормальность раствора,
mэ – эквивалентная масса
Так как кислотность основания Ca(OH)2 равна 2, то
![]()
![]()
3. (2,10-11) Определить концентрацию щелочи, если на титрование 5,86 мл её раствора пошло 7,28 мл 0,56 н раствора НСl
Решение:
Так как вещества взаимодействуют между собой в эквивалентных соотношениях, то растворы равной нормальности реагируют в равных объемах. При разных нормальностях объемы растворов реагирующих веществ обратно пропорциональны их нормальностям, т.е.:
![]()
![]()
4. (4,10-11) Сколько граммов PbO2 выделится на аноде при электролизе азотнокислого свинца током 0,15 А в течение 10 минут?
Решение:
По первому закону Фарадея количество вещества, выделяемого на электроде, прямо пропорционально количеству пропущенного электричества:
![]()
где ![]() – количество электричества
– количество электричества
![]() ,
,
где ![]() и
и ![]() сила тока и время соответственно,
сила тока и время соответственно,
![]() – постоянная Фарадея, 96500 Кл,
– постоянная Фарадея, 96500 Кл,
![]() .
.
![]()
![]()
5. (3-4,10) Условия съемки полярограмм в вольтамперометрии
Ответ:
Поскольку в вольтамперометрии один из электродов не поляризуется и для него потенциал остается постоянным, подаваемое на ячейку напряжение затрачивается на изменение потенциала только рабочего электрода. Если потенциал рабочего электрода измеряют относительно потенциала электрода сравнения, условно приняв последний за нуль, то в этом случае Е = Еа для рабочего микроанода и Е = - ЕК для рабочего микрокатода.
Таким
образом, регистрируемая вольтамперная кривая ![]() (полярограмма) отражает электрохимический процесс, происходящий только на
одном электроде.
(полярограмма) отражает электрохимический процесс, происходящий только на
одном электроде.
Если в
растворе присутствуют вещества, способные электрохимически восстанавливаться
или окисляться (так называемые деполяризаторы), то при наложении на ячейку
линейно меняющегося напряжения (скорость не превышает 200 мВ/мин) получаемая
кривая ![]() имеет форму волны, в отсутствие электрохимической реакции эта зависимость
линейна, как следует из закона Ома (рис. 5.1).
имеет форму волны, в отсутствие электрохимической реакции эта зависимость
линейна, как следует из закона Ома (рис. 5.1).
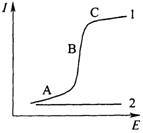
Рис. 5.1 Вольтамперная кривая в присутствии (1) и отсутствии (2) электрохимически активного соединения
При низких значениях потенциала (участок А на рис. 5.1), величина которого не достаточна для того, чтобы на рабочем микроэлектроде происходила электрохимическая реакция, через ячейку проходит очень незначительный остаточный ток, обусловленный прежде всего током заряжения двойного электрического слоя и присутствием в растворе электрохимически более активных, чем анализируемое вещество, примесей. При увеличении потенциала электрохимически активное вещество – деполяризатор вступает в электрохимическую реакцию на электроде и в результате этого ток резко возрастает (участок В). Это так называемый Фарадеевский ток. С ростом потенциала ток возрастает до некоторого предельного значения, оставаясь затем постоянным (участок С).
Предельный ток обусловлен тем, что в данной области потенциалов практически весь деполяризатор из при электродного слоя исчерпан в результате электрохимической реакции, а обеденный слой обогащается за счет диффузии деполяризатора из объема раствора. Скорость диффузии частиц деполяризатора в этих условиях контролирует скорость электрохимического процесса в целом. Такой ток называют предельным диффузионным.
Для того чтобы исключить электростатическое перемещение деполяризатора (миграцию) в поле электродов и понизить сопротивление ячейки, измерения, как было указано выше, проводят в присутствии большого избытка сильного электролита, называемого фоном. Являясь электрохимически индифферентным, вещество фонового раствора может вступать в химические реакции с определяемым веществом (часто это реакции комплексообразования). Иногда фоновый электролит играет роль буферного раствора. Например, при полярографическом определении ионов Cd2+, Zn2+, Ni2+, Со2+ в качестве фона используют аммиачный буферный раствор, который выполняет одновременно все вышеперечисленные функции.
Вольтамперная кривая описывается уравнением Гейровского-Ильковича
![]()
(знак "+" для анодного, "—" для катодного процессов),
где Е и Е1/2 — потенциал в любой точке полярографической волны и на половине ее высоты, соответственно;
п — число электронов, участвующих в электрохимической реакции;
![]() и
и ![]() — предельный диффузионный ток и ток для выбранного
значения напряжения Е.
— предельный диффузионный ток и ток для выбранного
значения напряжения Е.
Полярограмма содержит ценную аналитическую информацию:;потенциал полуволны Е1/2 является качественной характеристикой деполяризатора, в то время как предельный диффузионный ток линейно связан с концентрацией его в объеме раствора. Эта зависимость при использовании ртутного капающего микроэлектрода выражается уравнением Ильковича:
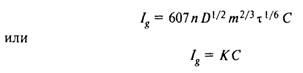
где D — коэффициент диффузии деполяризатора, см2/с; т —.масса ртути, вытекающей из капилляра за секунду, г/с; т — время жизни ртутной капли, с; С — объемная концентрация деполяризатора, моль/л. Для твердого микроэлектрода в отсутствие принудительно перемешивания раствора уравнение концентрационной зависимости тока принимает вид:
![]() ,
,
где S — площадь микроэлектрода, см2;
S — толщина диффузионного слоя, см.
Если в растворе присутствует несколько электрохимически активных соединений, на полярограмме наблюдается соответствующее число волн (рис. 5.2). Потенциалы полуволн и значения предельных диффузионных токов, зарегистрированных на такой полярограмме, совпадают с аналогичными величинами, полученными для растворов индивидуальных соединений такой же концентрации.
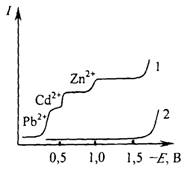
Рис. 5.2 Полярограмма раствора содержащего ионы Cd2+, Zn2+, Ni2+, Со2+ на фоне 1М раствора KCl (1): полярограмма фона (2).
Полярограммы могут быть искажены за счет полярографических максимумов — резкого возрастания тока выше его предельного значения с последующим спадом. Причины возникновения максимумов различны, и могут быть связаны с неравномерной поляризацией ртутной капли и тангенциальным движением ее поверхности, что приводит к дополнительному перемешиванию раствора. Такого рода максимумы можно устранить введением в полярографический раствор ПАВ: красителей (метиловый красный, фуксин и др.), высокомолекулярных соединений (агар-агар, желатин).
6. (4,10-11) Рассчитайте потенциал медного электрода, опущенного в 0,2 н раствор CuCl (н.у.)
Решение:
Стандартный потенциал пары Cu+/Cu равен 0,531 В. Можно пренебречь тем, что при нормальных условиях это значение изменится (так как это изменение будет не значительное).
Медный электрод, опущенный в раствор соли меди, является электродом первого рода, его потенциал зависит от природы потенциалопределяющей пары и концентрации катиона Сu+ (окисленной формы):
0,058
![]()
7. (5,10,11) Оптическая плотность 0,1 н раствора Cr3+ составляла А = 2,87. Измеренная в адекватных условиях оптическая плотность исследуемого раствора равнялась А = 4,25. Какова концентрация исследуемого раствора?
Решение:
![]() , где
, где ![]() молярный коэффициент поглощения;
молярный коэффициент поглощения; ![]() – толщина светопоглощающего слоя;
– толщина светопоглощающего слоя; ![]() – концентрация раствора.
– концентрация раствора.
Так как измерения проводились в адекватных условиях то можно записать
![]() ;
; ![]() и
и ![]() ; тогда
; тогда ![]()
![]()
8. (7,10) Сущность атомно-эмисссионного оптического спектрального анализа. Области его применения
Ответ:
Метод атомно-эмиссионной спектроскопии (АЭС) основан на термическом возбуждении свободных атомов или одноатомных ионов и регистрации оптического спектра испускания возбужденных атомов (рис. 8.1)
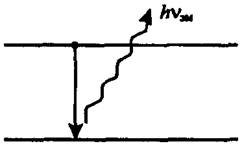
Рис. 8.1 Схема процесса лежащая в основе метода атомно-эмиссионной спектроскопии
Аналитическим
сигналом в АЭС служит интенсивность испускаемого излучения ![]() . Поскольку возбуждение атомов имеет термическую
природу, возбужденные и невозбужденные атомы находятся между собой в
термодинамическом равновесии, положение которого описывается законом
распределения Больцмана:
. Поскольку возбуждение атомов имеет термическую
природу, возбужденные и невозбужденные атомы находятся между собой в
термодинамическом равновесии, положение которого описывается законом
распределения Больцмана:
![]()
где N0 – число невозбужденных атомов;
g* и g0 – статистические веса возбужденного и невозбужденного состояния;
Е – энергия возбуждения;
к – постоянная Больцмана;
Т – абсолютная температура.
Таким образом, при постоянной температуре число возбужденных частиц N* прямо пропорционально числу невозбужденных частиц N0, т. е. фактически общему числу данных атомов N в атомизаторе (поскольку в реальных условиях атомно-эмиссионного анализа доля возбужденных частиц очень мала: N* << N0).
В свою очередь (при заданных условиях атомизации, определяемых конструкцией и режимом работы прибора и рядом других факторов), число атомов (N) в атомизаторе пропорционально концентрации определяемого элемента в пробе с.
Таким
образом, можно было бы ожидать, что между интенсивностью испускаемого излучения
![]() и концентрацией определяемого элемента с наблюдается
прямо пропорциональная зависимость. Однако на практике условия, обеспечивающие
эту зависимость, выполняются далеко не всегда. В общем случае зависимость
интенсивности излучения от концентрации нелинейная и может быть описана эмпирическим
уравнением
и концентрацией определяемого элемента с наблюдается
прямо пропорциональная зависимость. Однако на практике условия, обеспечивающие
эту зависимость, выполняются далеко не всегда. В общем случае зависимость
интенсивности излучения от концентрации нелинейная и может быть описана эмпирическим
уравнением
![]()
Это уравнение называется уравнением Ломакина-Шайбе. Оно является основным количественным соотношением атомно-эмиссионного анализа.
Коэффициент а в этом уравнении является сугубо эмпирической величиной, зависящей от условий процесса. Поэтому в АЭС решающее значение имеет правильный выбор условий атомизации и измерения аналитического сигнала, включая градуировку по образцам сравнения.
Области применения атомно-эмиссионный метода анализа
Качественный анализ. Атомно-эмиссионный метод позволяет одновременно зарегистрировать множество линий испускания. Поэтому АЭС является многоэлементным методом анализа. Это важнейшее достоинство метода позволяет успешно использовать его для идентификации элементов, содержащихся в пробе, для качественного анализа.
Из традиционных источников атомизации наиболее подходящим для качественного анализа является дуговой разряд. С одной стороны, температура дуги достаточна для атомизации и возбуждения большинства элементов. С другой стороны, поскольку температура дуги ниже, чем, например, искры или ИСП, то дуговые спектры существенно беднее линиями, что облегчает идентификацию. Основной недостаток дугового разряда – низкая стабильность – применительно к качественному анализу не играет существенной роли, поскольку для идентификации используют положение (длину волны) линии в спектре, а не ее интенсивность.
Для идентификации элементов используют в первую очередь наиболее интенсивные, так называемые «последние» линии (название связано с тем, что при уменьшении концентрации элемента эти линии исчезают в последнюю очередь). Чтобы идентификация была надежной, в спектре необходимо обнаружить несколько линий одного и того же элемента.
Количественный анализ. При количественном анализе методом АЭС можно использовать все основные способы градуировки – внешних стандартов (градуировочного графика), внутреннего стандарта и метод добавок. Целесообразность применения каждого способа зависит от характера возможных помех и природы анализируемого объекта. Так, метод добавок позволяет эффективно устранить косвенные мультипликативные погрешности, вызываемые главным образом физико-химическими помехами, однако против аддитивных спектральных помех – таких, как наложение спектральных линий, – он бессилен. Следует в то же время иметь в виду, что метод добавок легко реализуем технически только при анализе растворов (атомизаторы – главным образом пламя, ИСП), но не твердых проб (дуговой, искровой разряды). В любом случае при построении градуировочной зависимости следует стремиться к тому, чтобы все образцы, используемые для градуировки, были максимально адекватны анализируемому – как по валовому химическому составу, так и по физическому состоянию (последнее особо важно при анализе твердых проб).
9. (6,10-11) Вычислить длину волны линии лх, если расстояние её до линии железа л1 = 304,26 нм составляет 1,5 мм, а до линии железа л2=304,58 нм - 2,4 мм
Решение:
На рис. 9.1 представлено схематическое изображение небольшого участка спектра железа и спектра исследуемой пробы.
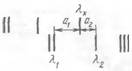
Рис. 9.1 Определение длины волны спектральной линии
Для определения длины волны заданной линии лх измеряют расстояние а1 от этой линии до ближайшей к ней линии спектра железа, длина волны которой л1 точно известна, и расстояние а2 от линии лх до другой линии с известной длиной волны л2. В небольшом спектральном интервале дисперсия прибора остается постоянной, поэтому можно записать пропорцию
![]()
После простых преобразований получаем формулу для расчета длины волны неизвестной линии:
![]()
![]()
10. (6,10-11) Определить длину волны неизвестной линии лх, если линии железа л1 = 327,4 нм и л2 = 328,8 нм находятся от нее на расстояниях соответственно 0,25 мм и 1,17 мм
Решение:
Все тоже, что и в предыдущей задачи:
![]() ;
;
![]()
11. (10) Нарисуйте принципиальную схему атомно-абсорбционного спектрометра и дайте пояснения к ней
Ответ:
На рис. 11.1 представлена принципиальная блок-схема ААС-спектрометра. Проба в нем вносится в атомизатор (например, пламя), где распадается до свободных атомов. Возбуждение атомов осуществляется потоком света УФ-видимой области, исходящего из лампы с полым катодом. Отсечение постороннего излучения и детектирование производятся при помощи таких устройств, как – монохроматор и ФЭУ (фотоэлектронный умножитель), соединенного с устройством отображения информации.
12. (10) Стандартные образцы состава
Ответ: Под стандартными образцами принято понимать образцы веществ или материалов, химический состав или физические свойства которых типичны для данной группы веществ (материалов), определены с необходимой точностью, отличаются высоким постоянством и удостоверены сертификатом.
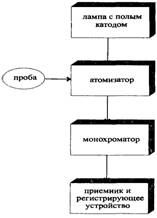
Рис. 11.1 Принципиальная схема атомно-абсорбционного спектрометра
Стандартные образцы используются для градуировки, поверки и калибровки химического состава и различных свойств материалов (механических, теплофизических, оптических и др.). Стандартные образцы как мера с установленной погрешностью (классом точности) применяются непосредственно для контроля качества сырья и промышленной продукции путем сличения. По существу стандартные образцы служат для поддержания единства измерений, т.е. являются средствами измерений.
В основе классификации стандартных образцов лежат:
• разновидность характеристики, по которой проводится аттестация стандартного образца;
• метод анализа (сличения) объектов контроля со стандартным образцом;
• агрегатное состояние самого стандартного образца как материла (вещества);
• метрологическое назначение.
Согласно этой классификации стандартные образцы подразделяют по первому признаку на образцы свойств материалов (веществ) и образцы состава материалов (веществ); по второму признаку различают стандартные образцы для химического, рентгеновского, спектроскопического и других видов анализа; по третьему признаку – стандартные образцы в твердом, жидком и газообразном состоянии; по метрологическому назначению (четвертый признак) – стандартные образцы для градуировки, поверки, контроля качества вещества и т.д.
Особо важное значение имеет категория стандартных образцов для установления чистоты веществ. Понятие особо чистых веществ связано с производством многих материалов современной техники, медицины и т.д. Стандартные образцы подвергаются специальным испытаниям, по результатам которых они получают свидетельства (сертификат) и вносятся в государственный реестр стандартных образцов, а он в свою очередь является составной частью (разделом) Государственного реестра средств измерений. В сертификате стандартного образца обязательно указывается срок годности, поскольку практически все вещества и материалы со временем изменяются вследствие воздействия факторов окружающей среды на их свойства. А от этого зависит достоверность результатов измерений.
К настоящему времени опубликованы данные более чем о 3,5 млн. веществ и материалов, что характеризует значимость такого средства измерений, как стандартные образцы состава и свойств веществ и материалов.
В России действует Государственная служба стандартных образцов (ГССО) в составе НПО "ВНИИМ им. Д.И. Менделеева". Главная цель этой службы – обеспечение любой организации, нуждающейся в проведении контроля качества своей продукции с помощью стандартных образцов, образцами и изготовление новых образцов по заявкам заинтересованных юридических лиц.
13. (6,10) Рассчитайте разрешающую способность оптического спектрометра, если спектральные линии с длинами волн 273,85 нм и 273,80 нм не перекрываются
Решение:
Разрешающей способностью спектрального прибора называют его способность давать раздельное изображение двух спектральных линий с близкими длинами волн.
Количественной
характеристикой разрешающей способности прибора является отношение ![]() , где
, где ![]() – интервал, в котором линии
– интервал, в котором линии ![]() и
и ![]() наблюдаются раздельно, а
наблюдаются раздельно, а ![]() – средняя длина волны.
– средняя длина волны.
![]()
![]()
![]()
14. (10) Рассчитайте случайную ошибку определения серы в стали, если в ходе анализа были получены следующие данные: 0,0123; 0,0125; 0,0121; 0,0123; 0,0122 % S
Решение:
Наличие грубых ошибок (промахов) оцениваем по Q-критерию. Располагаем экспериментальные данные в порядке возрастания величин 0,0121, 0,0122, 0,0123, 0,0123, 0,0125.
Предполагаем, что значения 0,0121 и 0,0125 являются результатом случайной ошибки.
Рассчитаем критерий Q для этих величин по формуле
![]()
где ![]() – размах варьирования
– размах варьирования ![]() – разница между двумя крайними значениями в ряду
величин
– разница между двумя крайними значениями в ряду
величин ![]() (расположенных в порядке возрастания)
(расположенных в порядке возрастания) ![]() – подозрительно выделяющееся значение;
– подозрительно выделяющееся значение; ![]() – соседнее с ним значение.
– соседнее с ним значение.
![]()
![]()
Для ![]() и
и ![]() табличное значение
табличное значение ![]() .
. ![]() и
и ![]() , поэтому грубая ошибка в измерениях 0,0121 и 0,0125
отсутствует.
, поэтому грубая ошибка в измерениях 0,0121 и 0,0125
отсутствует.
![]()
Находим стандартное отклонение:
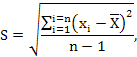
где ![]() – единичное отклонение, т. е. отклонение отдельного
измерения от среднего арифметического;
– единичное отклонение, т. е. отклонение отдельного
измерения от среднего арифметического; ![]() – число степеней свободы.
– число степеней свободы.
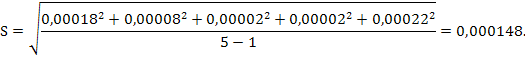
Стандартное отклонение среднего результата равно:
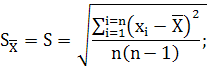
![]()
Вычисляем доверительный интервал, принимая
![]()
![]() ;
;
![]()
![]()
![]()
Оцениваем
еще раз наличие случайных ошибок по критерию ![]() :
:
![]()
Сравнивая
величины ![]() и
и ![]() , видим, что ни одно из отклонений от среднего не
выходит за пределы
, видим, что ни одно из отклонений от среднего не
выходит за пределы ![]() . Следовательно величины
. Следовательно величины ![]() не содержат грубых ошибок.
не содержат грубых ошибок.
Так как выборка не содержит грубых ошибок то ее можно обрабатывать с применением методов математической статистики.
Тогда,
интервал, в котором с вероятностью ![]() лежит истинное значение, равен
лежит истинное значение, равен
![]()
т. е. ![]() % S –
это и есть оценка случайной ошибки.
% S –
это и есть оценка случайной ошибки.
15. (10) Что такое концентрационная чувствительность методики анализа?
Ответ:
Концентрационная чувствительность метода или методики определяется тем минимальным количеством вещества, которое можно обнаруживать или определять данным методом, по данной методике. На рис. 15.1 приведена относительная характеристика чувствительности некоторых методов. Нижняя граница определяемого содержания демонстрирует возможности метода и наилучший результат, достигаемый при определении ряда веществ.
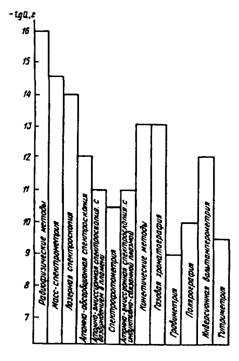
Рис.
15.1 Нижние границы определяемых содержаний компонентов ![]() для некоторых методов анализа
для некоторых методов анализа
Сопоставляя концентрационную чувствительность различных методов и оценивая примерное содержание компонента в образце, химик выбирает тот или метод анализа. Например, для определения содержания натрия в силикатных породах используют гравиметрический метод, позволяющий определять миллиграммовые и более высокие количества натрия; для определения микрограммовых количеств того же элемента в растениях и биологических образцах животного происхождения – метод пламенной фотометрии; для определения натрия в воде особой чистоты (нано- и пикограммовые количества) – метод лазерной спектроскопии.